Welcome Back! See Course tab for Course Materials, see YouTube tab for livestream recordings
FCS Clean Up for GitHub
Author
David Rach
Published
May 23, 2026
Getting Started
GitHub has a file-size limit for individual files (~ 5MB). Since the smallest raw .fcs file in the dataset being used for the TRU-OLS walkthrough was 23 MB, the dataset needed to be cleaned up and downsampled before inclusion as an example dataset.
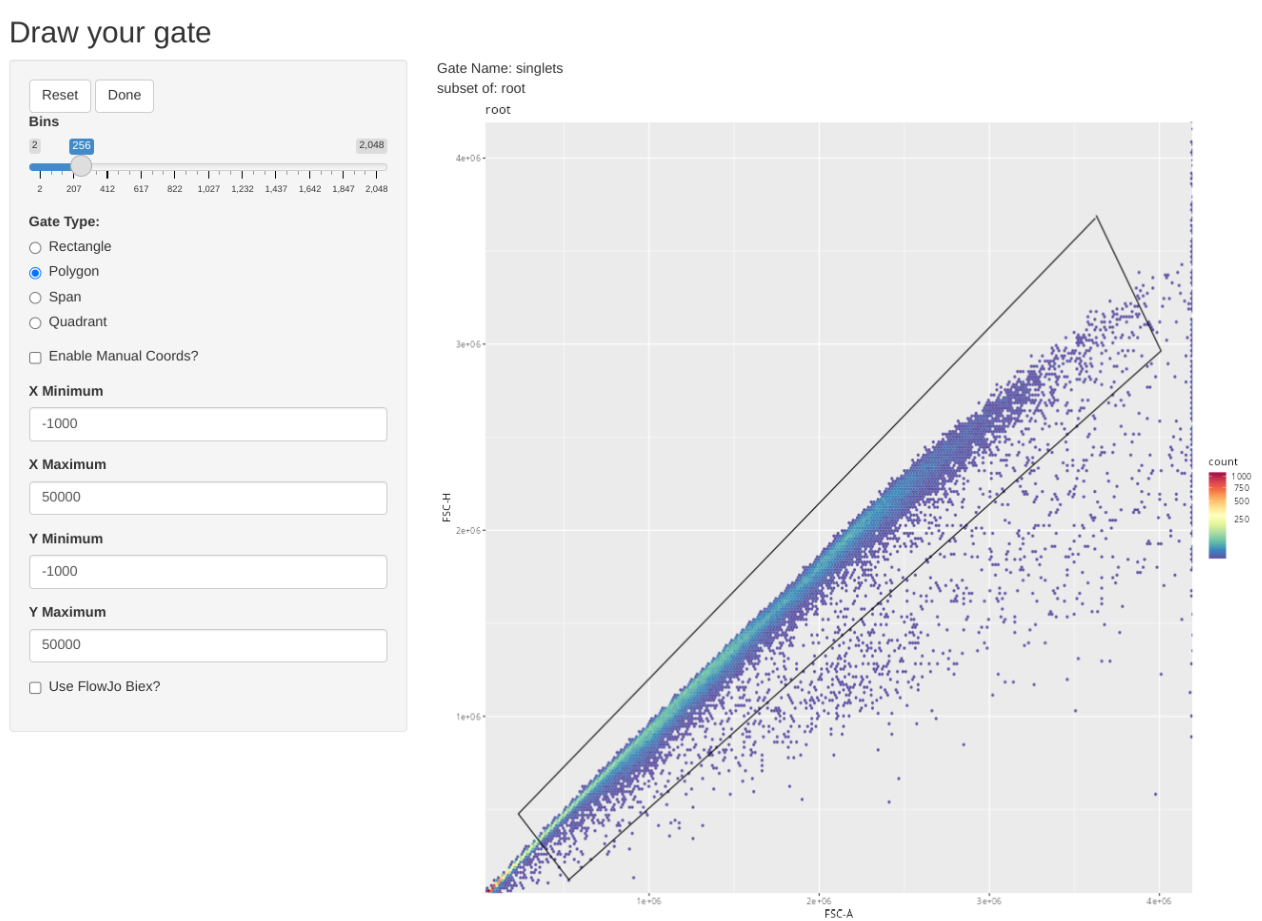
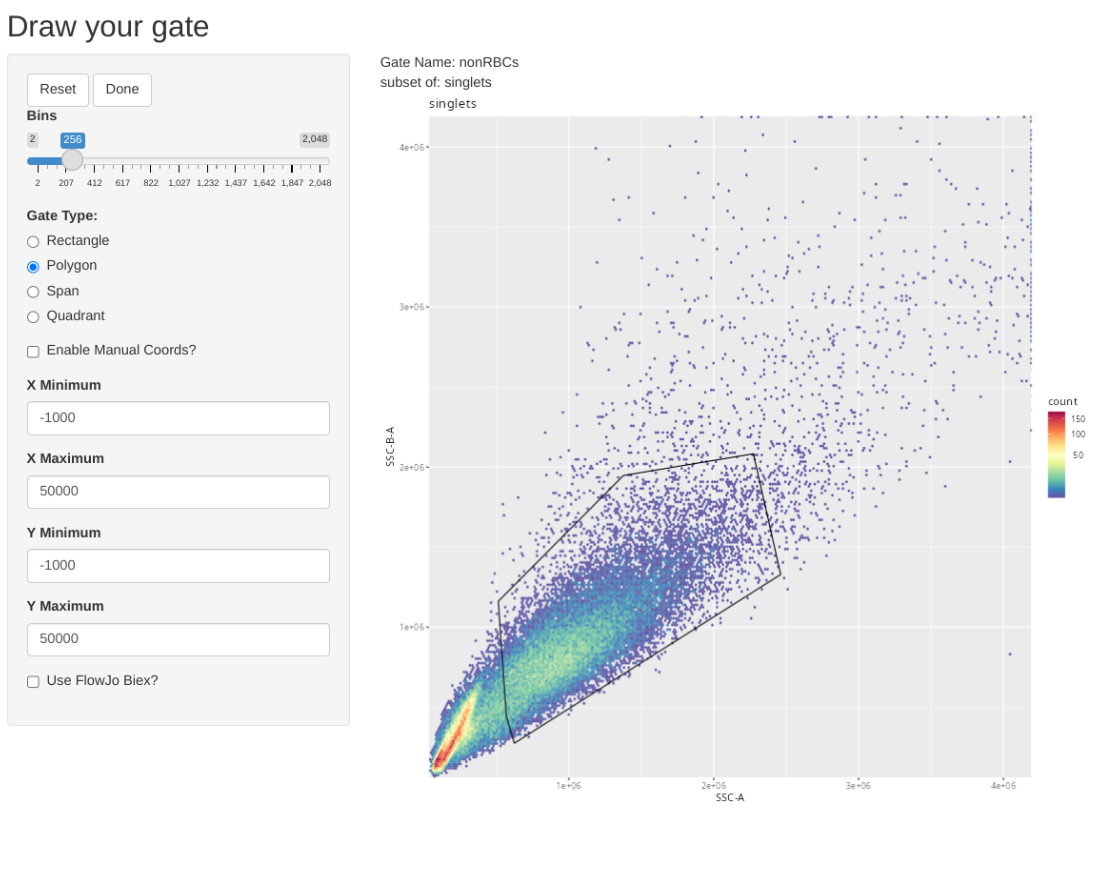
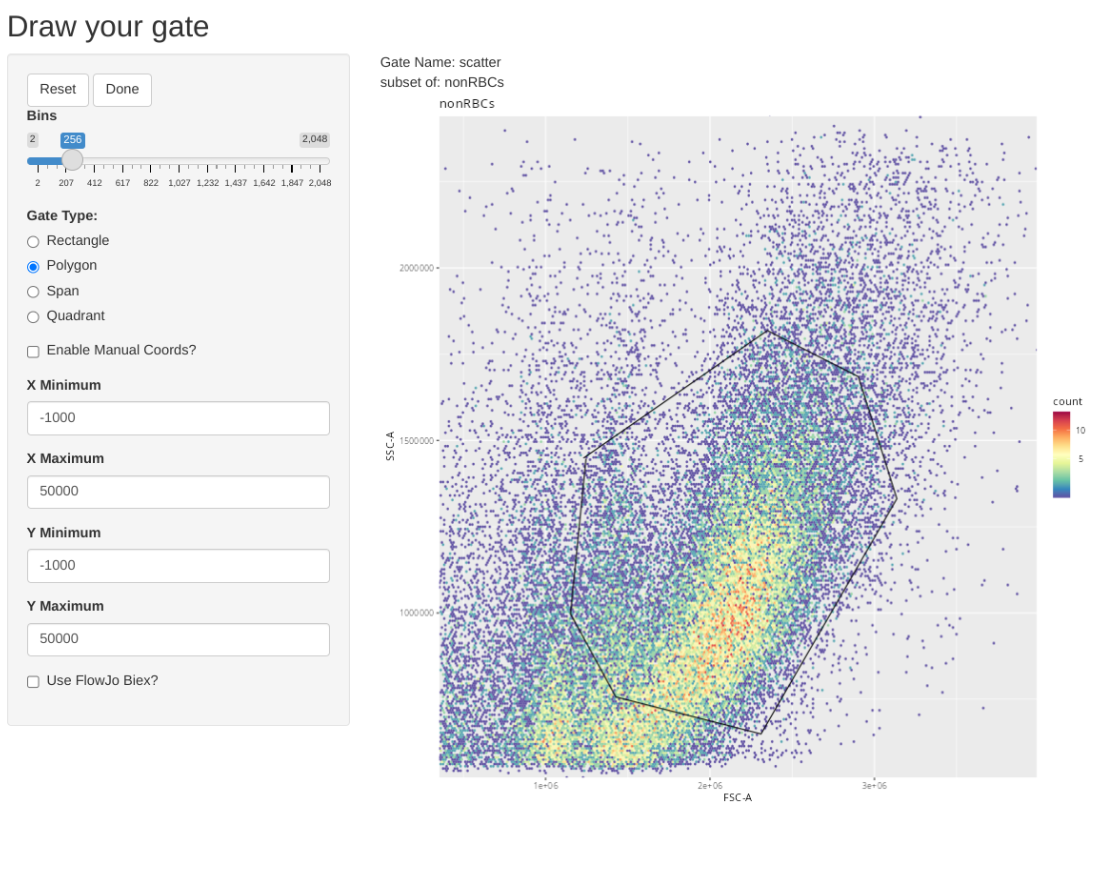
Consequently we will first gate for singlets, non-RBCs and lymphocyte scatter. Once this has been accomplished, we will downsample for 17500 of the present cells, which will result in being just under the 5 MB limit when using 5-laser Cytek Aurora raw .fcs files.
As always, start by loading the required R packages via the library() call.
library(flowWorkspace)
As part of improvements to flowWorkspace, some behavior of
GatingSet objects has changed. For details, please read the section
titled "The cytoframe and cytoset classes" in the package vignette:
vignette("flowWorkspace-Introduction", "flowWorkspace")
library(flowGate)
Loading required package: ggcyto
Loading required package: ggplot2
Loading required package: flowCore
Loading required package: ncdfFlow
Loading required package: BH
Registered polygon_gate
Registered span_gate
library(dplyr)
Attaching package: 'dplyr'
The following object is masked from 'package:ncdfFlow':
filter
The following object is masked from 'package:flowCore':
filter
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
library(purrr)
Next, activate the Downsampling() function that was created as part of Week 10 of the course.
RFolder <-file.path("course", "community", "TRU-OLS", "R") # For Interactive# RFolder <- file.path("R") # For Quarto RenderingMyFunctions <-list.files(RFolder, full.names=TRUE)purrr::walk(.x=MyFunctions, .f=source)
With this done, specify your input file.path and output file.paths
As we will be working with raw .fcs files today, we do not need to transform. If we did need to, we would use the following code (modifying the arguments to match our own instrument settings).
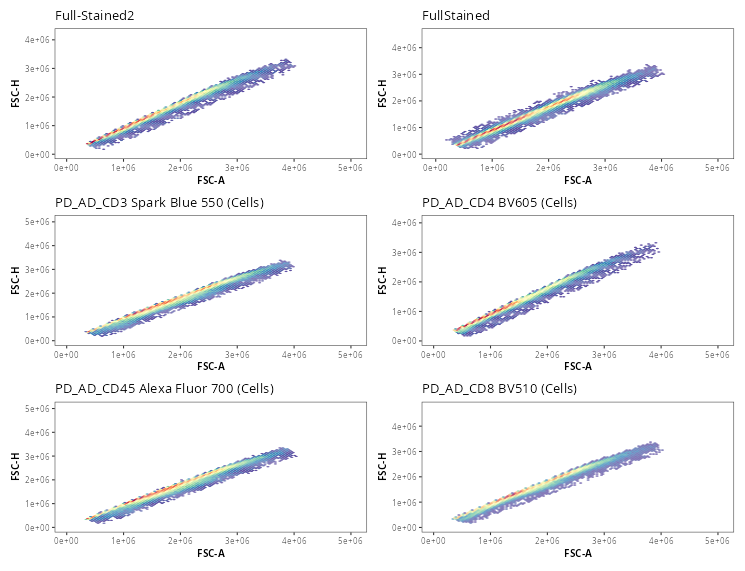
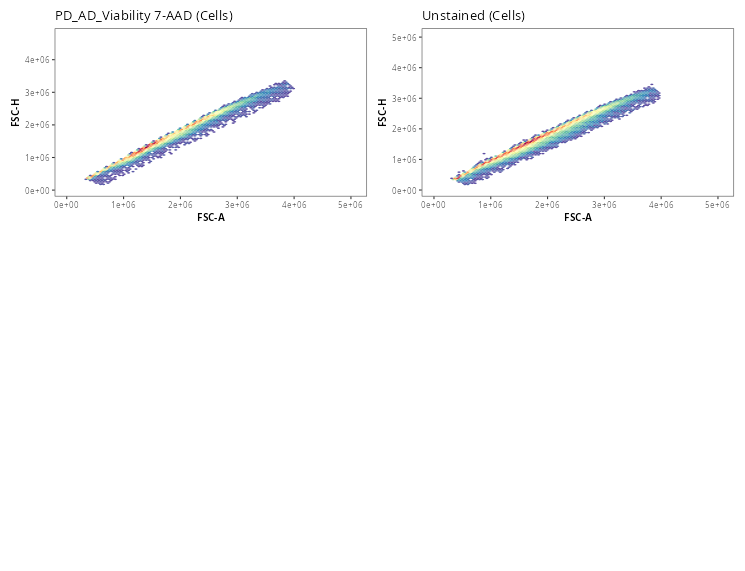
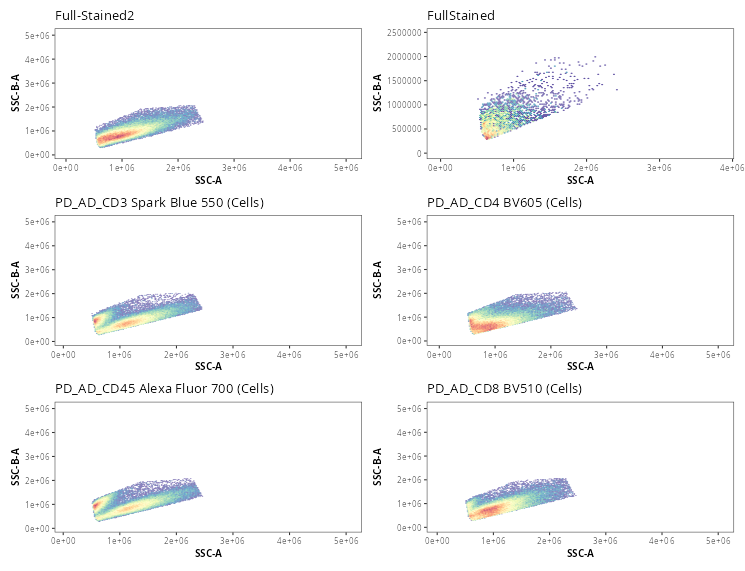
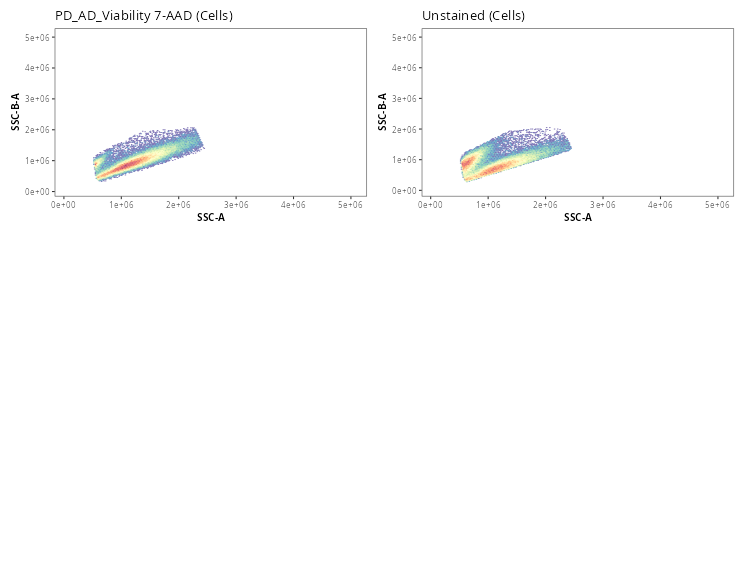
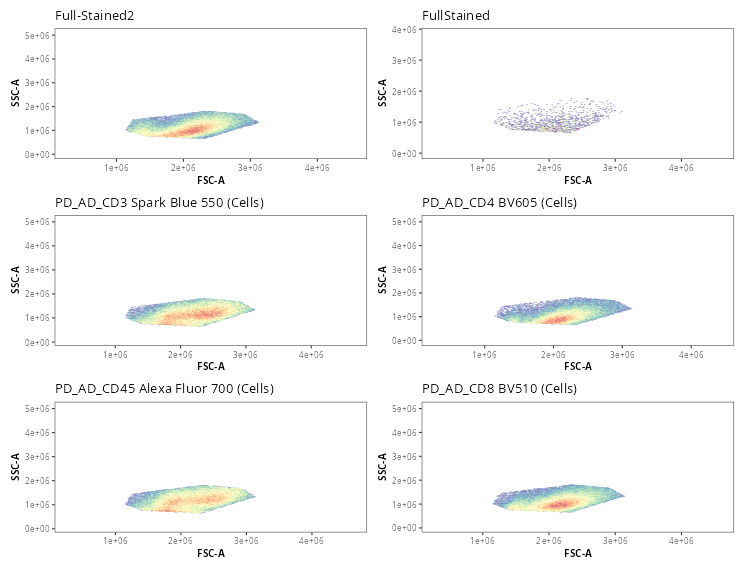
Once the gates are drawn, we can quickly validate that the gate placements will suffice for what we are trying to accomplish today. We can use the Utility_UnityPlots() from the Luciernaga package to compare the same gate across all specimens
For a raw .fcs file from a 5-laser Cytek Aurora, downsampling to 17500 cells puts us just under the 5 MB limit. Seeing as we have enough cells, lets proceed to export out the downsampled .fcs files to our output folder.
The resulting files are now under 5 MB. I proceeded to remove the original ones from the “data” folder, and replaced them with the files from the outputs folder. These were then transferred to GitHub for use in this walk-through example.
To return to where you were in the TRU-OLS walk-through, click here