Week 7: For this seventh session, we take a closer look at the raw values of the data within our .fcs files, and explore the various ways to transform (ie. scale) flow cytometry data in R to better visualize “positive” and “negative populations”.
There exist several commonly used transformations for Flow Cytometry data. Similarly, for Mass Cytometry data, the arsinh transformation is the most frequently applied.
Regardless of method, the goal remains to rescale the data in a way that allows for better interpretation. A misapplied transformation can be dangerous for an analysis, so don’t just accept the default. Always ensure that what your transformed data visually makes sense. Similarly, this approach is shared in many of the exploratory data analsyis approaches we will encounter later on during the course.
Additionally, due to near perfect timing, Felix Marsh-Wakefield did a good overview of transformation for CytoBites YouTube channel earlier this week that is worth checking out.
Walk Through
ImportantHousekeeping
As we do every week, on GitHub, sync your forked version of the CytometryInR course to bring in the most recent updates. Then within Positron, pull in those changes to your local computer.
For YouTube walkthrough of this process, click here
After setting up a “Week07” project folder, copy over the contents of “course/06_Visualizing/data” to that folder. This will hopefully prevent merge issues next week when attempting to pull in new course material. Once you have your new project folder organized, remember to commit and push your changes to GitHub to maintain remote version control.
If you encounter issues syncing due to the Take-Home Problem merge conflict, see this walkthrough. The updated homework submission protocol can be found here
Load Libraries
This week, we will extensively be using the flowWorkspace package, as we learn how to build out our own GatingSet objects to include transformations. As we do so, we will need to visualize our underlying data, to ensure that the transformartions applied were correct for our datasets. Consequently, we will be using the ggcyto package extensively throughout the day. Therefore, it makes sense to go ahead and attach both packages to our local environment from the start.
library(flowWorkspace)
As part of improvements to flowWorkspace, some behavior of
GatingSet objects has changed. For details, please read the section
titled "The cytoframe and cytoset classes" in the package vignette:
vignette("flowWorkspace-Introduction", "flowWorkspace")
library(ggcyto)
Loading required package: ggplot2
Loading required package: flowCore
Loading required package: ncdfFlow
Loading required package: BH
Separating SFC and MC fcs files
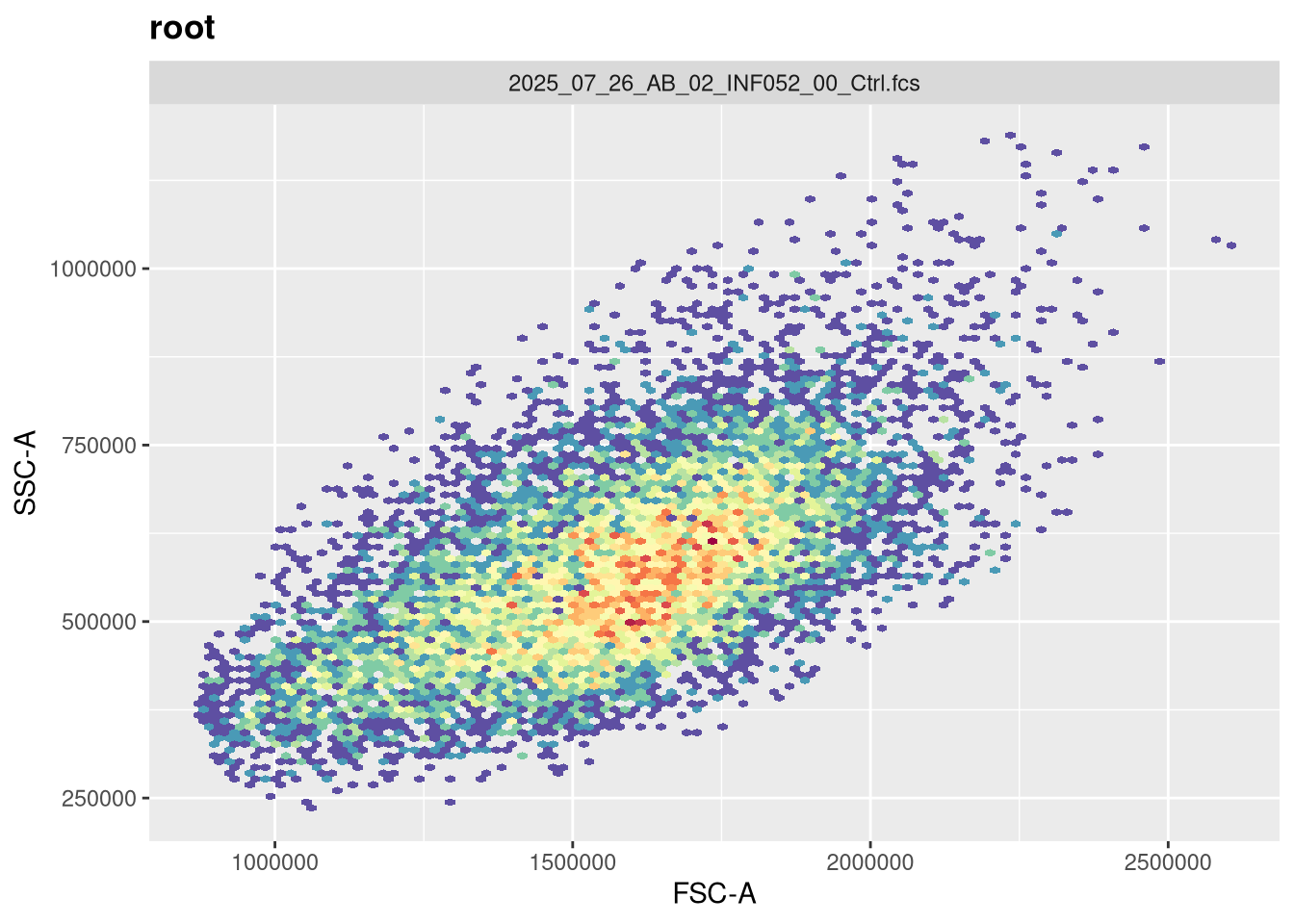
We will be using two cytometry datasets this week. For the Spectral Flow Cytometry dataset, we will be re-using the 6 unmixed .fcs file first shared during Week 05. This time around, rather than bring them from a FlowJo.wsp into a GatingSet via the CytoML package, we will be building the GatingSet from scratch using the flowWorkspace package. These .fcs file names share the “2025” portion in the name, which we will use to filter them from the list.files() vector list.
For the Mass Cytometry dataset, we will be using 3 .fcs files that I retrieved from ImmPort for the Study SDY2739 (Clinical Immunity to Malaria Involves Epigenetic Reprogramming of Innate Immune Cells). The .fcs file names for these files share the “G1_2” portion in the name, which we will use to filter them from the list.files() vector list.
#StorageLocation <- file.path("course", "07_Transformations", "data") # When interactively writing the code StorageLocation <-file.path("data") #When Quarto Renderingfcs_files <-list.files(StorageLocation, ".fcs", full.names=TRUE)SFC_files <- fcs_files[stringr::str_detect(fcs_files, "2025")]MC_files <- fcs_files[stringr::str_detect(fcs_files, "G1_2")]
Lets start by working with the SFC dataset. First off, let’s double check that we have just the SFC files (as flowWorkspace will throw an error if the .fcs files passed contain different number of fluorophore/metal columns)
Circling back to where we left off during Week 05, we will pass these file path locations to load_cytoset_from_fcs() to load them into a GatingSet object.
In the current form, the metadata is kinda empty, only containing rownames and the name column. However, we can notice that within the existing names, we have some useful information present between the underscores that we might be able to extract out and append as additional metadata colums.
Lets first off retrieve the metadata by assigning to its own object/variable
As you can see, it is just a data.frame style object that we have encountered in the past. We can therefore use dplyr package functions to help us tidy the metadata to extract the additional information that we may need in the future. Let’s go ahead and load in dplyr
library(dplyr)
Attaching package: 'dplyr'
The following object is masked from 'package:ncdfFlow':
filter
The following object is masked from 'package:flowCore':
filter
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
library(stringr)
And then create a new column for Condition so that we can better evaluate how the various extraction functions work, and also not risk messing up the underlying data while testing out how to use several new functions.
Testing <- Metadata |>mutate(Condition = name)
RegEx
Glancing at the current names, we can notice that if we wanted to extract the Condition (Ctrl, PPD, SEB), we would need to remove all character values up the final underscore (_), and before the “.fcs”.
Within base R, two functions, sub() and gsub() are most frequently encountered when attempting to extract out a particular pattern. These rely on providing a regular expression (RegEx) pattern to match, and a replacement value to swap in its place (which can be left empty by providing a ““).
Consequently, if we were using sub(), our first step to replace everything up through the last underscore would be the RegEx expression “.*_”
Please be advised, even with years of practice, I still will struggle to remember the proper RegEx syntax code to extract out a pattern from a long complicated character string. This is one use case where having a LLM provide a narrowly focused answer can be quite useful. Prompt example:
“I am working in R, using sub(), I want to keep everything after the last _ : 2025_07_26_AB_02_INF052_00_Ctrl.fcs”
In addition to sub() and gsub() in base R, we can also achieve a similar outcome using the stringr package from the tidyverse, using the str_extract() function (after investigating the correct RegEx style syntax). To interpret this RegEx-inspired hieroglyphic, note that “^” is used for the start of a string, and “$” for the end of the string. So we are extracting the pattern after the underscore “^_“, but keeping the contents before the final .fcs
Having emerged from the metadata, let’s return to focusing on our SFC dataset.
SFC_GatingSet
A GatingSet with 6 samples
colnames
When working with .fcs files that we have not worked with recently, it is often good to go ahead and check to see what fluorophores/markers are present at the start. This is also useful as different cytometer platforms have slightly different naming conventions. We can use colnames()
As we can see from the output, most of the fluorophores have an “-A” appended to the end, although the FSC and SSC parameters feature area, height and width (“-A”, “-H”, “-W”) respectively.
markernames
If we wanted to retrieve the markers corresponding to the individual fluorophores, rather than attempting the breaking into the S4 object as we did during Week 03, we can take advantage of the markernames() function provided by the developers.
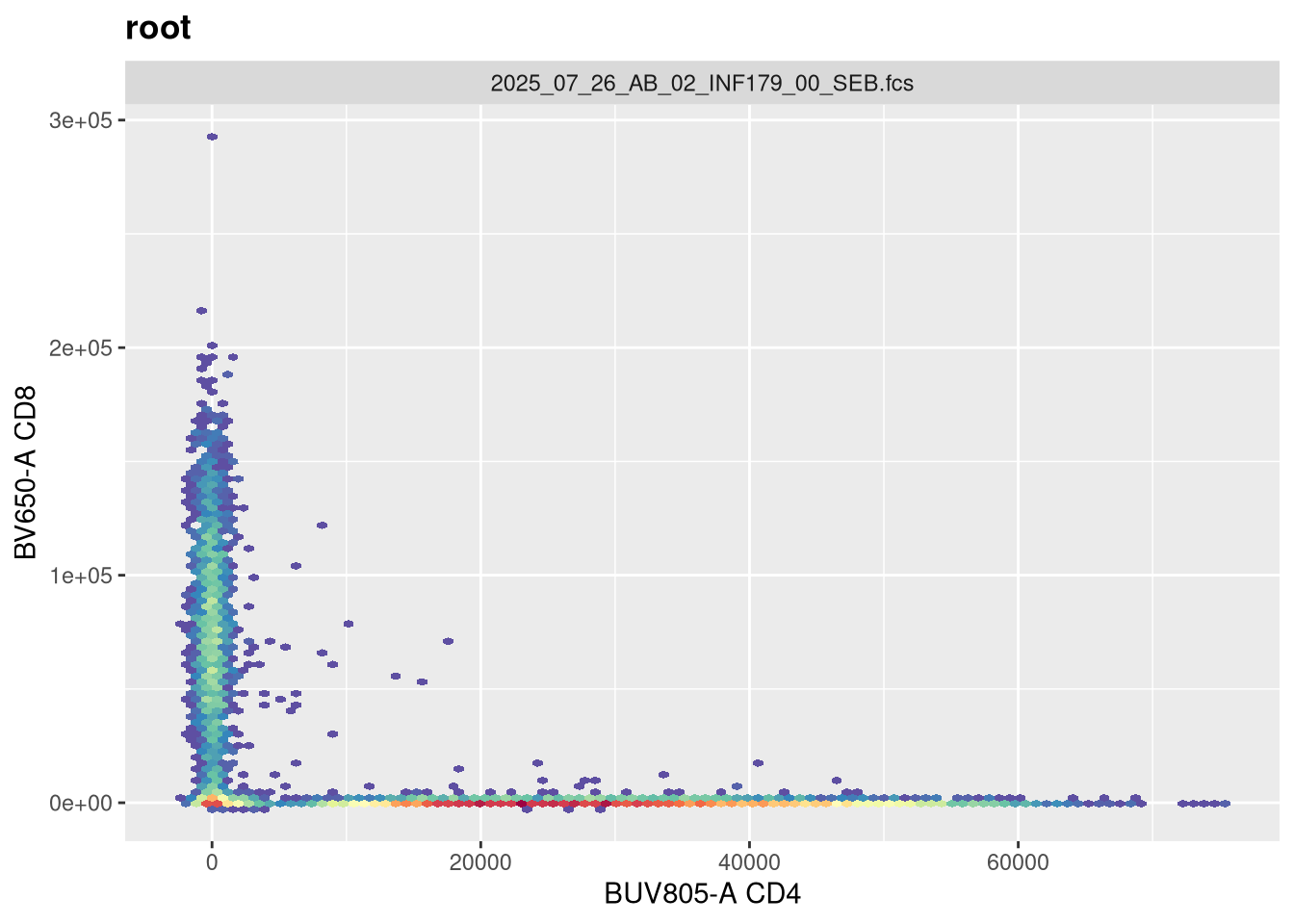
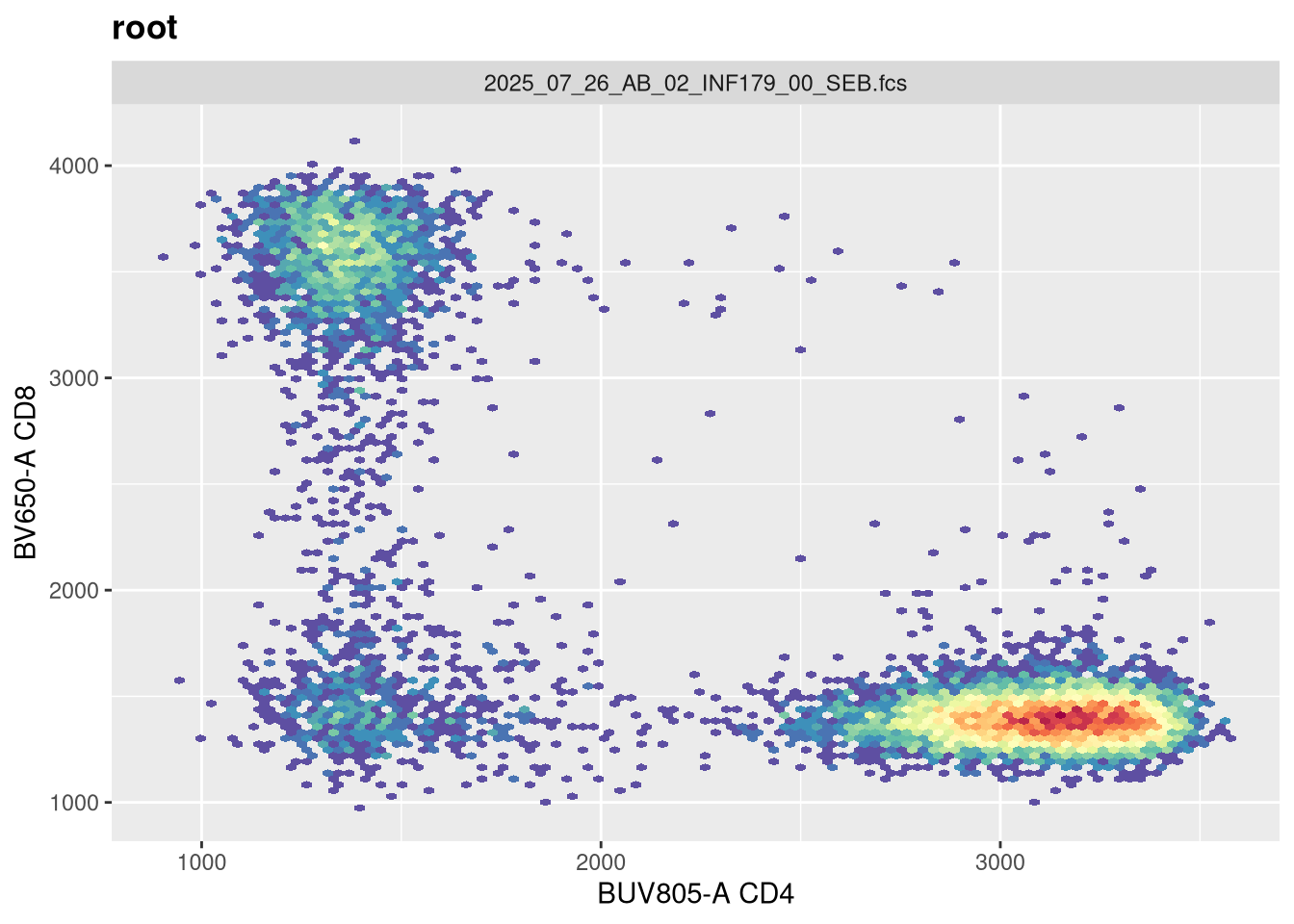
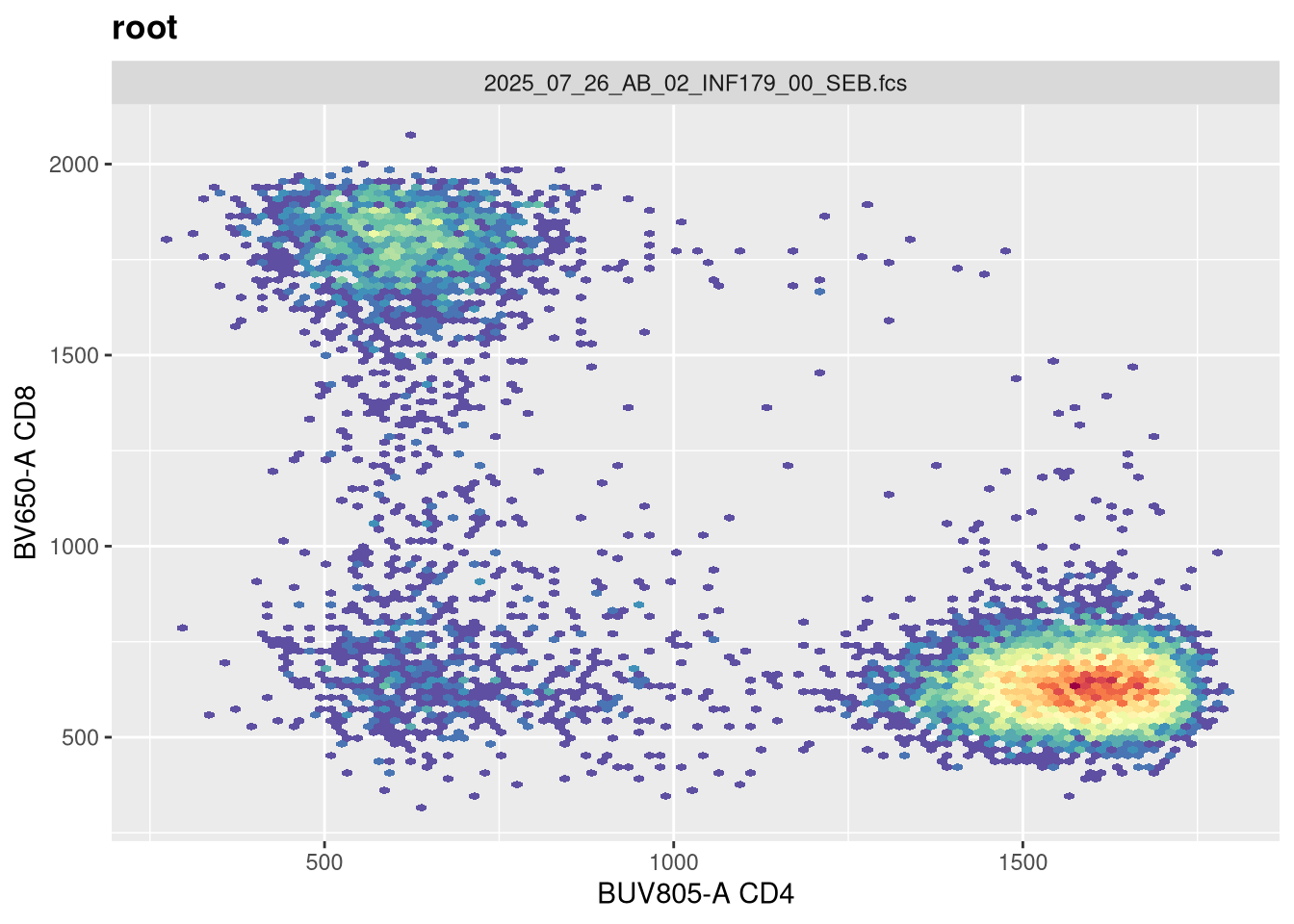
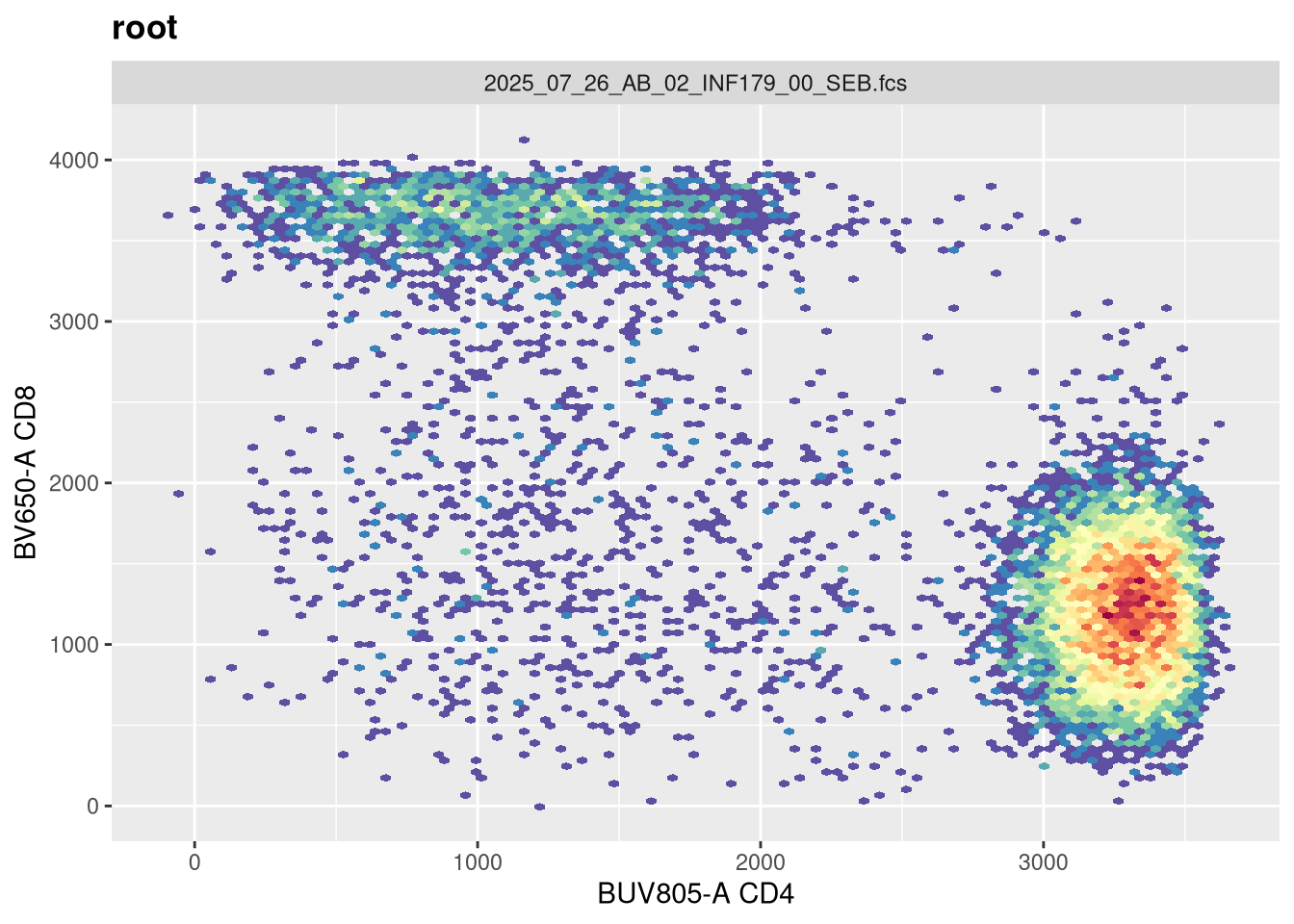
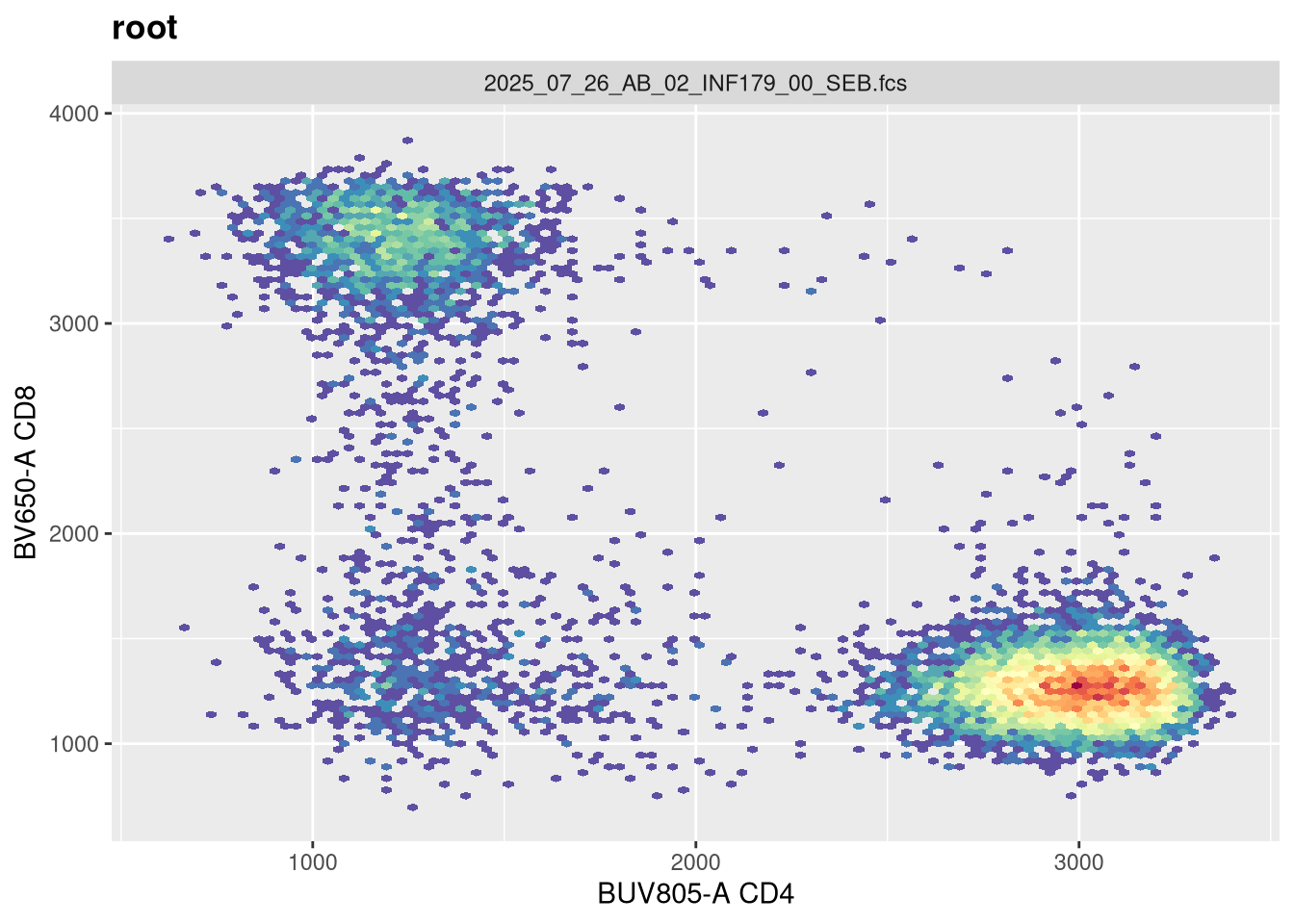
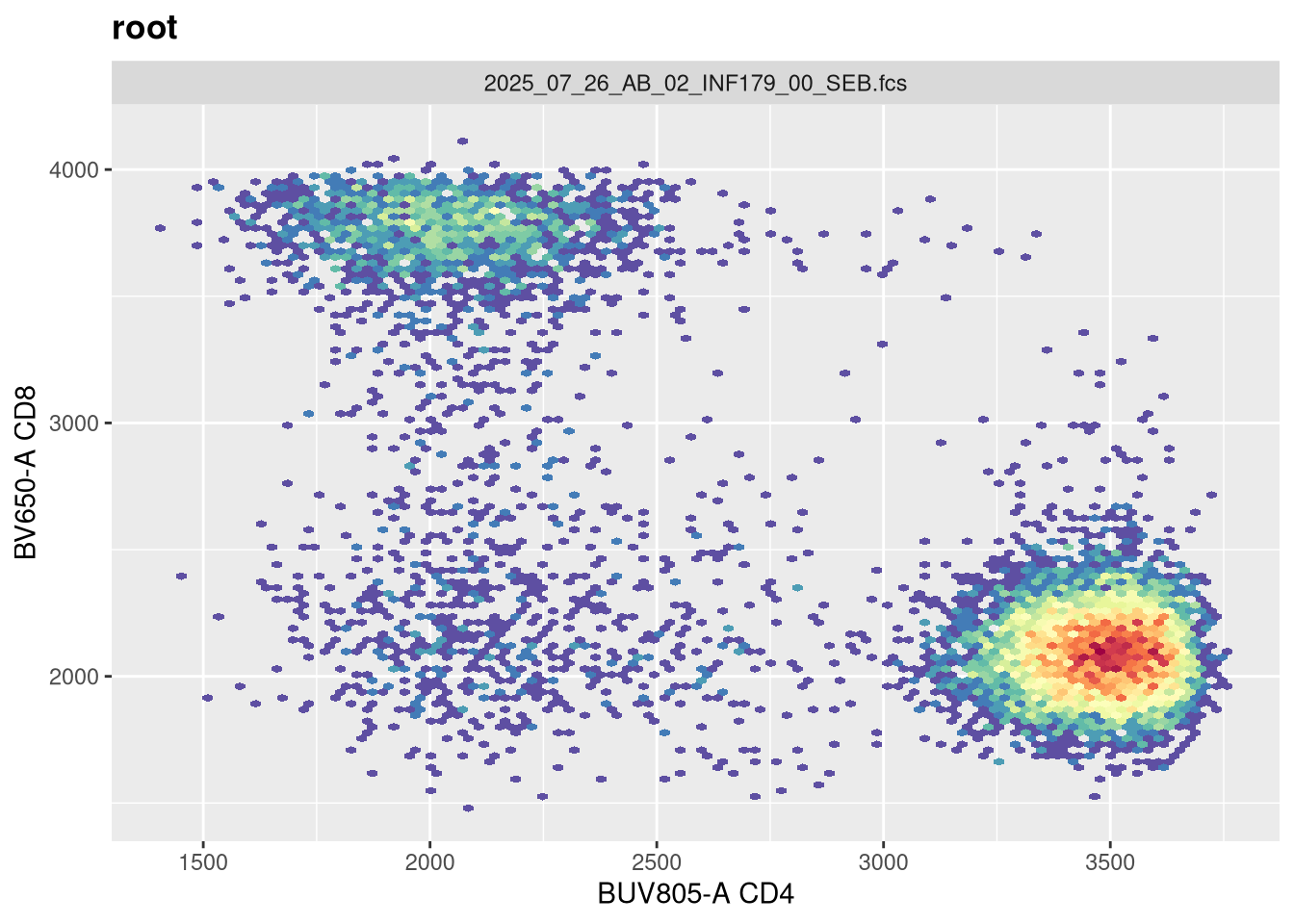
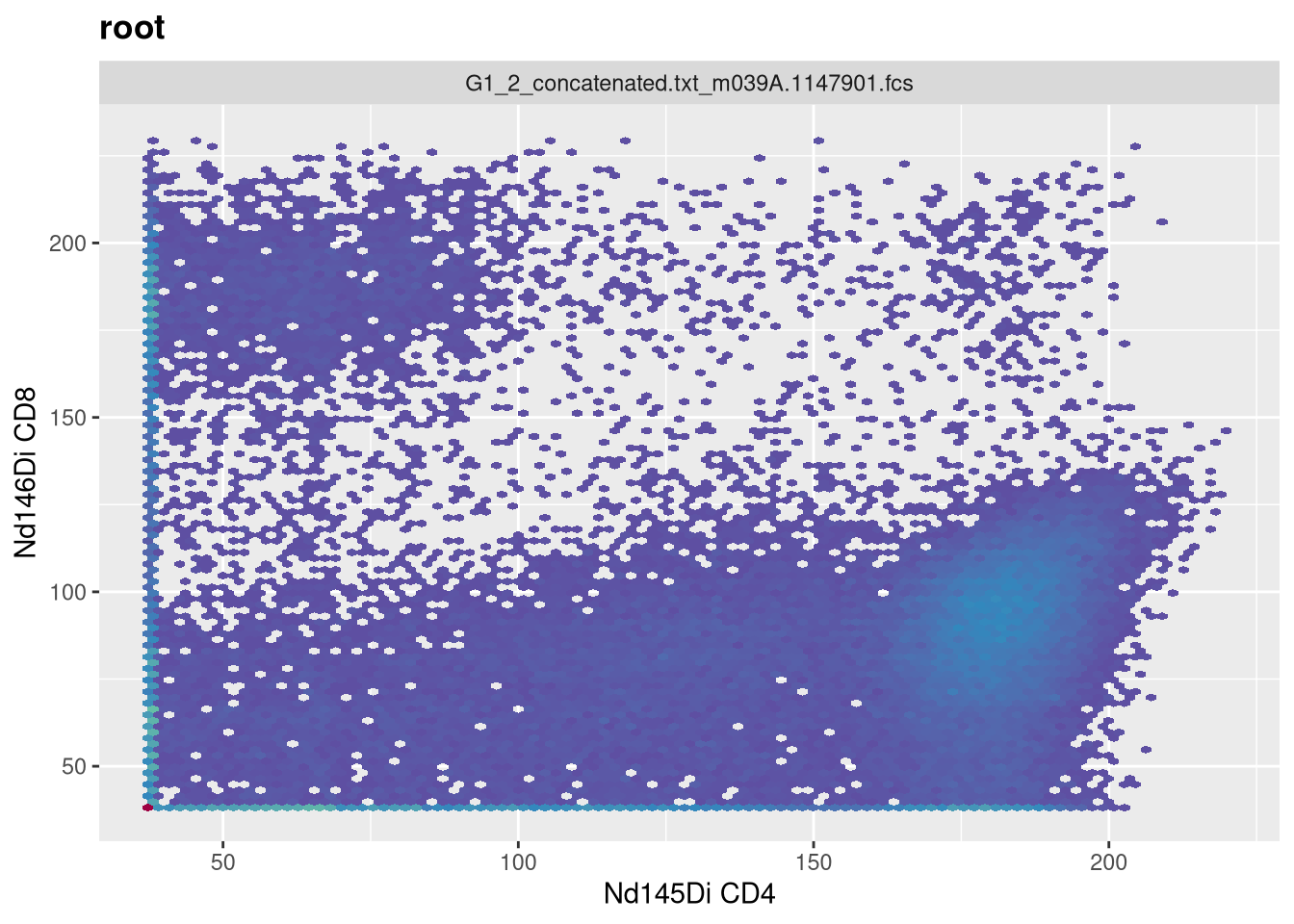
As we can see, the scale currently appears linear, with a wide stretch of positive values for both CD4 and CD8 T cells. By contrast, you may remember the transformed version brought for this same .fcs file brought in from FlowJo via CytoML resembled the following
If we use the gh_get_transformations function, we can see that there are currently no transformations applied to the GatingSet.
gh_get_transformations(SFC_GatingSet[[1]])
list()
Transformers
flowWorkspace provides several default transforms (with the option to create your own). The functions in question return transformer objects, which are list that collect the required information for the transformations that are subsequently applied.
Logicle <-logicle_trans()str(Logicle)
List of 9
$ name : chr "logicle"
$ transform :function (x)
$ inverse :function (x)
$ d_transform : NULL
$ d_inverse : NULL
$ breaks :function (x)
$ minor_breaks:function (b, limits, n)
$ format :function (x)
$ domain : num [1:2] -Inf Inf
- attr(*, "class")= chr "transform"
List of 9
$ name : chr "flowJo_biexp"
$ transform :function (x, deriv = 0)
..- attr(*, "type")= chr "biexp"
..- attr(*, "parameters")=List of 5
.. ..$ channelRange: int 4096
.. ..$ maxValue : num 262144
.. ..$ neg : num 0
.. ..$ pos : num 4.5
.. ..$ widthBasis : num -10
$ inverse :function (x, deriv = 0)
..- attr(*, "type")= chr "biexp"
..- attr(*, "parameters")=List of 5
.. ..$ channelRange: int 4096
.. ..$ maxValue : num 262144
.. ..$ neg : num 0
.. ..$ pos : num 4.5
.. ..$ widthBasis : num -10
$ d_transform : NULL
$ d_inverse : NULL
$ breaks :function (x)
$ minor_breaks:function (b, limits, n)
$ format :function (x)
$ domain : num [1:2] -Inf Inf
- attr(*, "class")= chr "transform"
Asinh <-flowjo_fasinh_trans()str(Asinh)
List of 9
$ name : chr "flowJo_fasinh"
$ transform :function (x)
$ inverse :function (x)
$ d_transform : NULL
$ d_inverse : NULL
$ breaks :function (x)
$ minor_breaks:function (b, limits, n)
$ format :function (x)
$ domain : num [1:2] -Inf Inf
- attr(*, "class")= chr "transform"
AsinhGML <-asinhtGml2_trans()str(AsinhGML)
List of 9
$ name : chr "asinhtGml2"
$ transform :function (x)
$ inverse :function (x)
$ d_transform : NULL
$ d_inverse : NULL
$ breaks :function (x)
$ minor_breaks:function (b, limits, n)
$ format :function (x)
$ domain : num [1:2] -Inf Inf
- attr(*, "class")= chr "transform"
As always, it can be worthwhile to first check the help documentation, to investigate the various arguments that can be used within the setup.
Column names to be transformed
While having the transformation parameters is one component, the other is the fluorophores that are to be transformed. Recalling the colnames() function, we can see we have the following fluorophores for this panel.
We will only need to apply transformations to fluorophores, not to FSC, SSC or Time parameters. We will therefore need to remove these from the list. One way would be to use [] index method, combining a ! and stringrstr_detect() to remove for values that would be shared by those parameters
Now that we have the fluorophore columns identified, using the transformerList() function we can combine them with the Transformer object we previously created.
List of 3
$ BUV395-A:List of 9
..$ name : chr "flowJo_biexp"
..$ transform :function (x, deriv = 0)
.. ..- attr(*, "type")= chr "biexp"
.. ..- attr(*, "parameters")=List of 5
.. .. ..$ channelRange: int 4096
.. .. ..$ maxValue : num 262144
.. .. ..$ neg : num 0
.. .. ..$ pos : num 4.5
.. .. ..$ widthBasis : num -10
..$ inverse :function (x, deriv = 0)
.. ..- attr(*, "type")= chr "biexp"
.. ..- attr(*, "parameters")=List of 5
.. .. ..$ channelRange: int 4096
.. .. ..$ maxValue : num 262144
.. .. ..$ neg : num 0
.. .. ..$ pos : num 4.5
.. .. ..$ widthBasis : num -10
..$ d_transform : NULL
..$ d_inverse : NULL
..$ breaks :function (x)
..$ minor_breaks:function (b, limits, n)
..$ format :function (x)
..$ domain : num [1:2] -Inf Inf
..- attr(*, "class")= chr "transform"
$ BUV563-A:List of 9
..$ name : chr "flowJo_biexp"
..$ transform :function (x, deriv = 0)
.. ..- attr(*, "type")= chr "biexp"
.. ..- attr(*, "parameters")=List of 5
.. .. ..$ channelRange: int 4096
.. .. ..$ maxValue : num 262144
.. .. ..$ neg : num 0
.. .. ..$ pos : num 4.5
.. .. ..$ widthBasis : num -10
..$ inverse :function (x, deriv = 0)
.. ..- attr(*, "type")= chr "biexp"
.. ..- attr(*, "parameters")=List of 5
.. .. ..$ channelRange: int 4096
.. .. ..$ maxValue : num 262144
.. .. ..$ neg : num 0
.. .. ..$ pos : num 4.5
.. .. ..$ widthBasis : num -10
..$ d_transform : NULL
..$ d_inverse : NULL
..$ breaks :function (x)
..$ minor_breaks:function (b, limits, n)
..$ format :function (x)
..$ domain : num [1:2] -Inf Inf
..- attr(*, "class")= chr "transform"
$ BUV615-A:List of 9
..$ name : chr "flowJo_biexp"
..$ transform :function (x, deriv = 0)
.. ..- attr(*, "type")= chr "biexp"
.. ..- attr(*, "parameters")=List of 5
.. .. ..$ channelRange: int 4096
.. .. ..$ maxValue : num 262144
.. .. ..$ neg : num 0
.. .. ..$ pos : num 4.5
.. .. ..$ widthBasis : num -10
..$ inverse :function (x, deriv = 0)
.. ..- attr(*, "type")= chr "biexp"
.. ..- attr(*, "parameters")=List of 5
.. .. ..$ channelRange: int 4096
.. .. ..$ maxValue : num 262144
.. .. ..$ neg : num 0
.. .. ..$ pos : num 4.5
.. .. ..$ widthBasis : num -10
..$ d_transform : NULL
..$ d_inverse : NULL
..$ breaks :function (x)
..$ minor_breaks:function (b, limits, n)
..$ format :function (x)
..$ domain : num [1:2] -Inf Inf
..- attr(*, "class")= chr "transform"
str(MyBiexTransform$`APC-A`)
List of 9
$ name : chr "flowJo_biexp"
$ transform :function (x, deriv = 0)
..- attr(*, "type")= chr "biexp"
..- attr(*, "parameters")=List of 5
.. ..$ channelRange: int 4096
.. ..$ maxValue : num 262144
.. ..$ neg : num 0
.. ..$ pos : num 4.5
.. ..$ widthBasis : num -10
$ inverse :function (x, deriv = 0)
..- attr(*, "type")= chr "biexp"
..- attr(*, "parameters")=List of 5
.. ..$ channelRange: int 4096
.. ..$ maxValue : num 262144
.. ..$ neg : num 0
.. ..$ pos : num 4.5
.. ..$ widthBasis : num -10
$ d_transform : NULL
$ d_inverse : NULL
$ breaks :function (x)
$ minor_breaks:function (b, limits, n)
$ format :function (x)
$ domain : num [1:2] -Inf Inf
- attr(*, "class")= chr "transform"
As you can notice, for each of the fluorophores we specified, the transformer object with the desired parameters has now been added as its own list entry.
The final step is to then apply it to the GatingSet
transform(SFC_GatingSet, MyBiexTransform)
A GatingSet with 6 samples
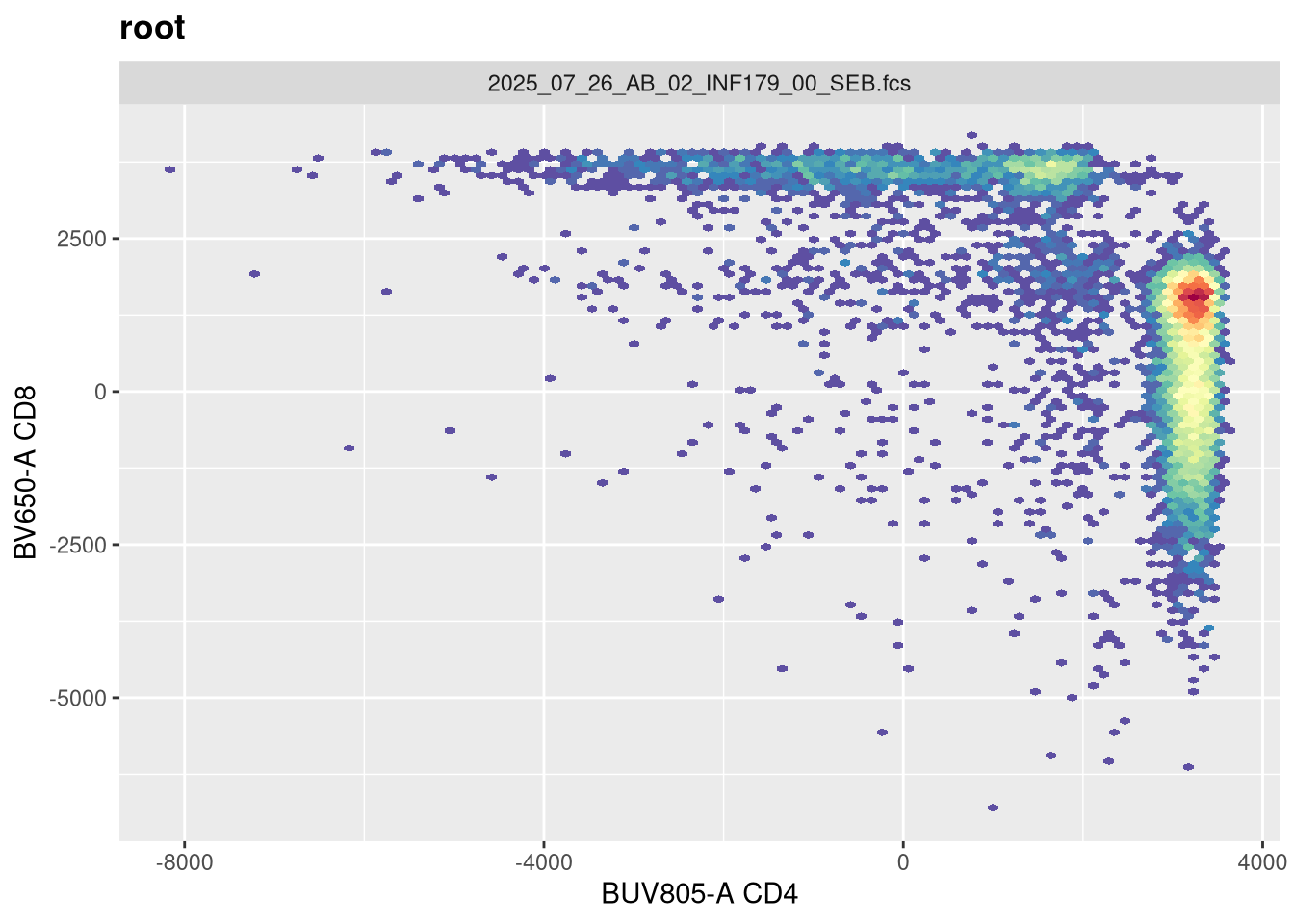
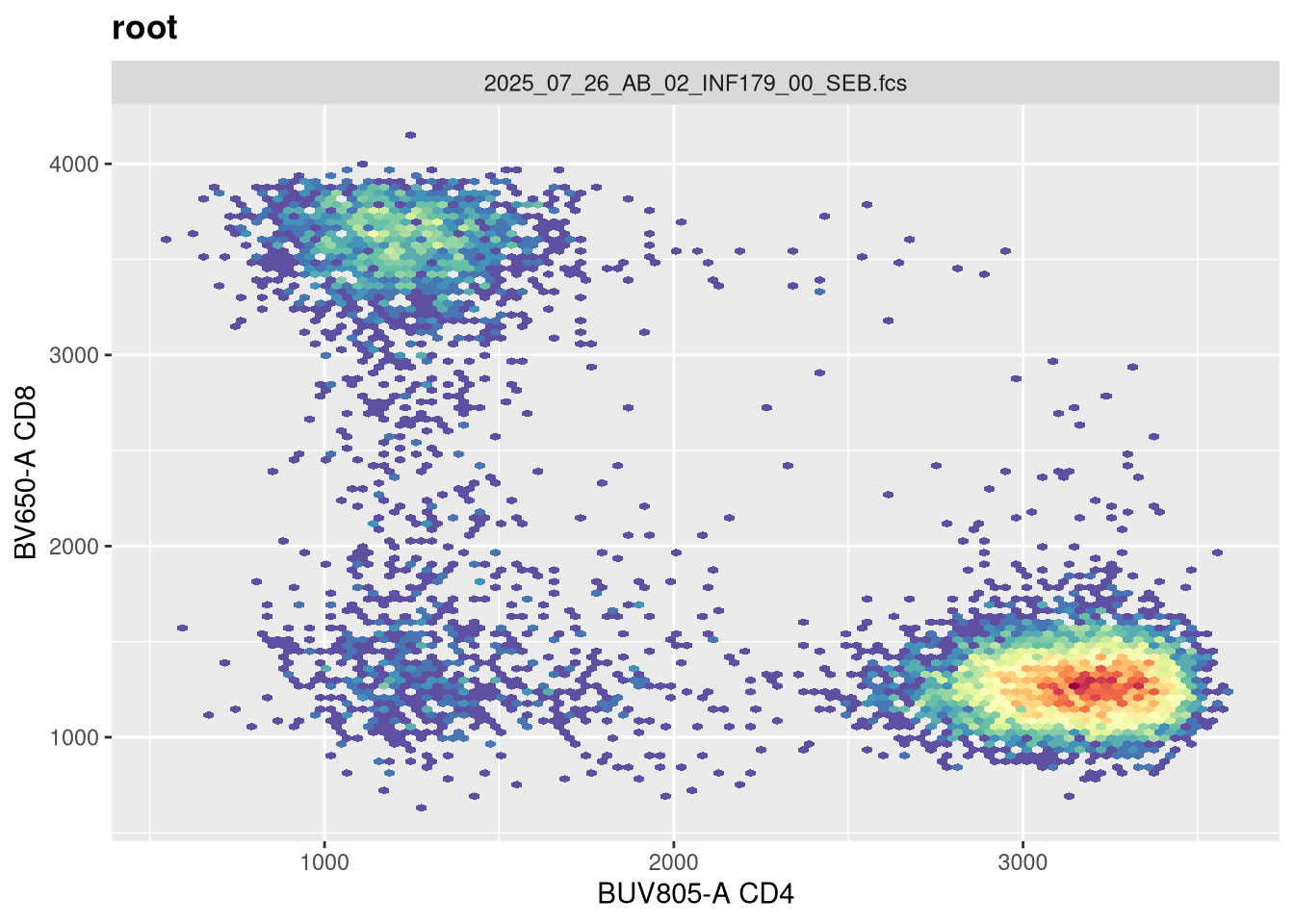
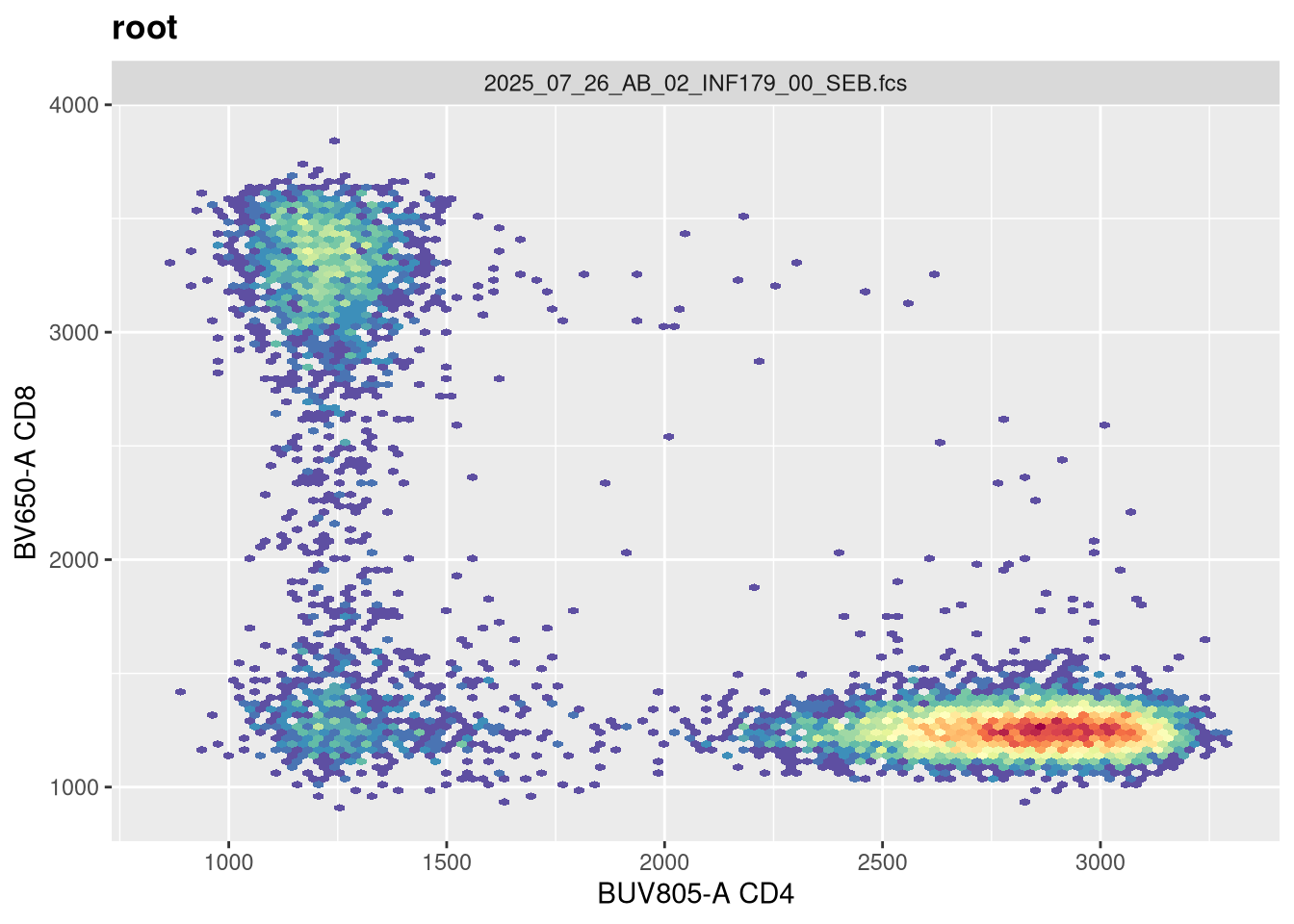
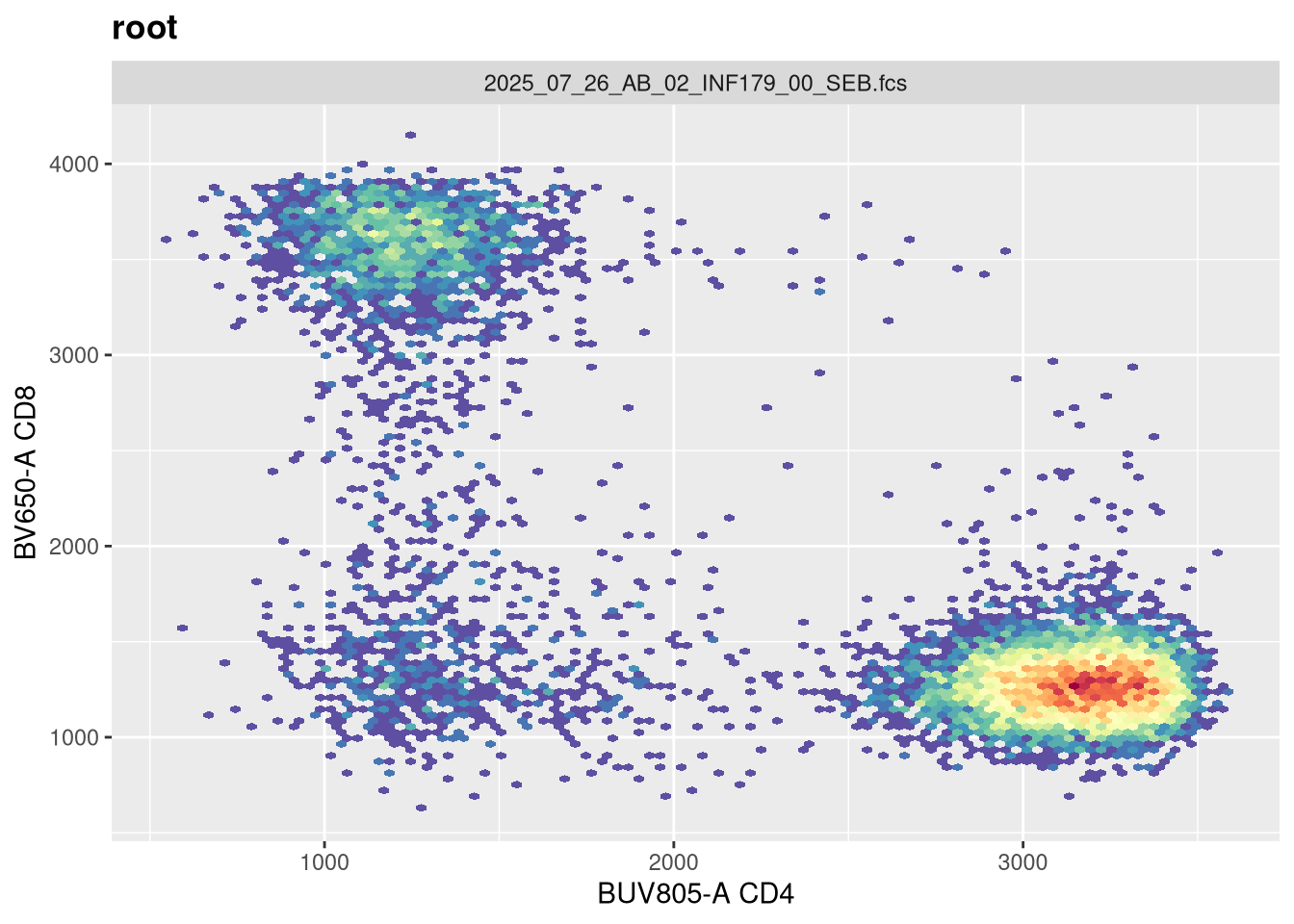
If we now replot without having provided any arguments, we get the following:
As we can see, applying a default transformation to a random .fcs file is not quite the way to go. We will need to look up some of the specific parameter arguments that need to be provided. Going back to our transformer function
?flowjo_biexp_trans
In this case, the help documentation doesn’t provide much immediately. Part of this is that flowjo_biexp_trans() appears to be a wrapper function, which passes arguments on to the flowJoTrans() function. If we look this one up
?flowJoTrans
Under usage, we can see what the default options and arguments are.
Transformation Arguments
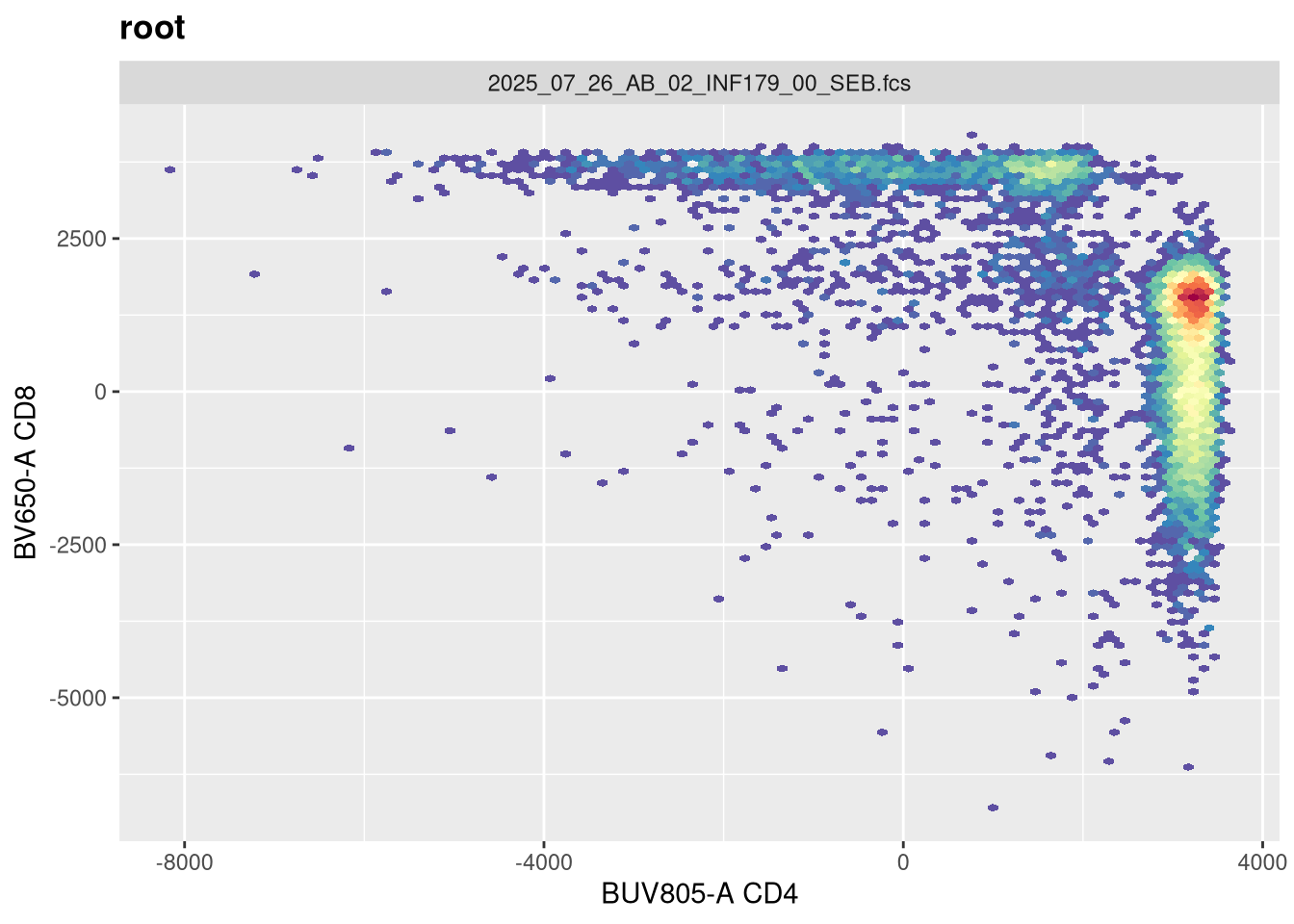
Lets evaluate what effect changing each of these arguments in has on visualizing our .fcs file. Our current visual is
We will also need a way to validate whether they are being updated in the background. We can revisit the output of gh_get_transformations() function from earlier, and specify just the first specimen, and the BUV805 fluorophore
function (x, deriv = 0)
{
deriv <- as.integer(deriv)
if (deriv < 0 || deriv > 3)
stop("'deriv' must be between 0 and 3")
if (deriv > 0) {
z0 <- double(z$n)
z[c("y", "b", "c")] <- switch(deriv, list(y = z$b, b = 2 *
z$c, c = 3 * z$d), list(y = 2 * z$c, b = 6 * z$d,
c = z0), list(y = 6 * z$d, b = z0, c = z0))
z[["d"]] <- z0
}
res <- stats:::.splinefun(x, z)
if (deriv > 0 && z$method == 2 && any(ind <- x <= z$x[1L]))
res[ind] <- ifelse(deriv == 1, z$y[1L], 0)
res
}
<bytecode: 0x55af21c73228>
<environment: 0x55af2c22a270>
attr(,"type")
[1] "biexp"
attr(,"parameters")
attr(,"parameters")$channelRange
[1] 4096
attr(,"parameters")$maxValue
[1] 262144
attr(,"parameters")$neg
[1] 0
attr(,"parameters")$pos
[1] 4.5
attr(,"parameters")$widthBasis
[1] -10
width
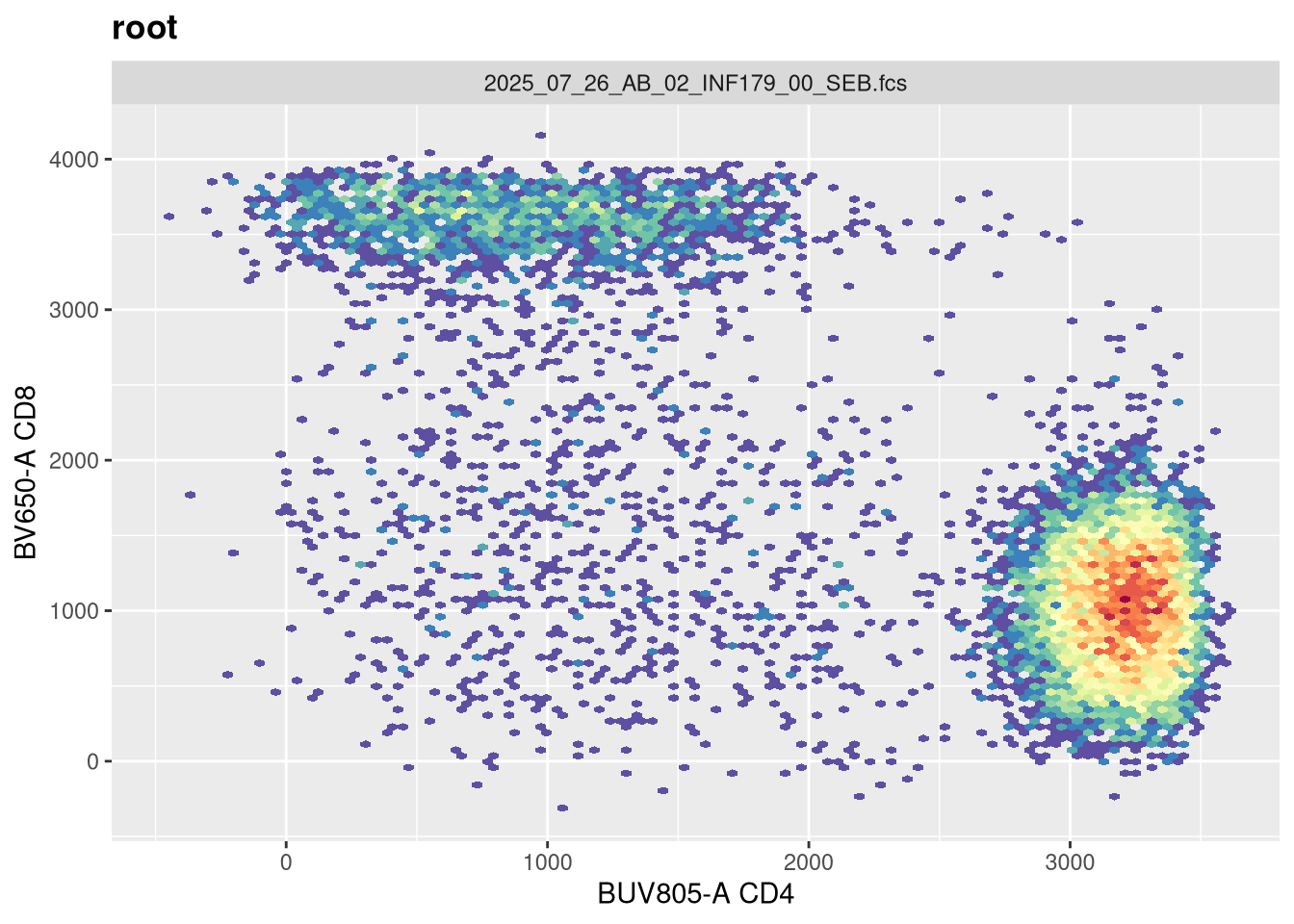
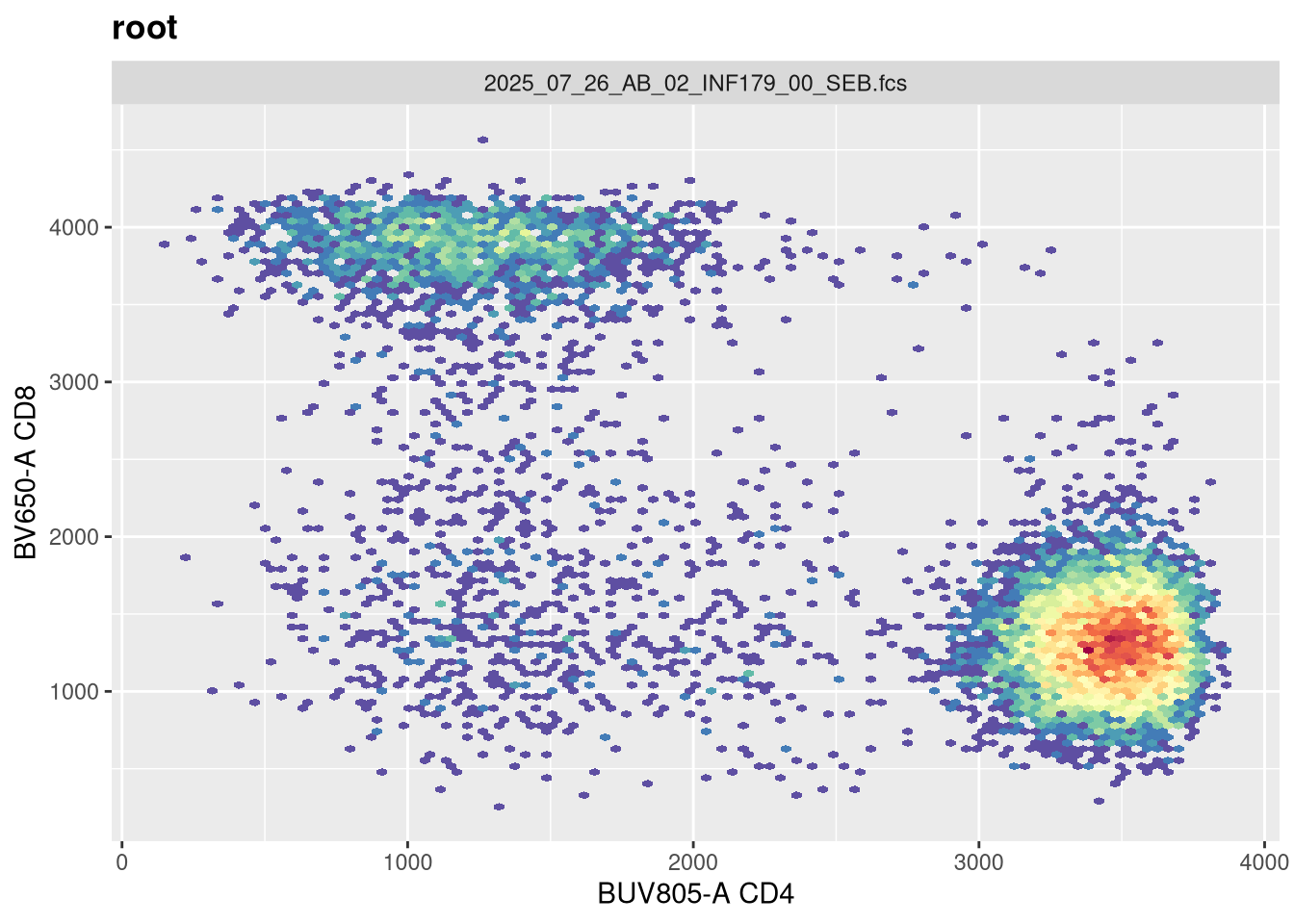
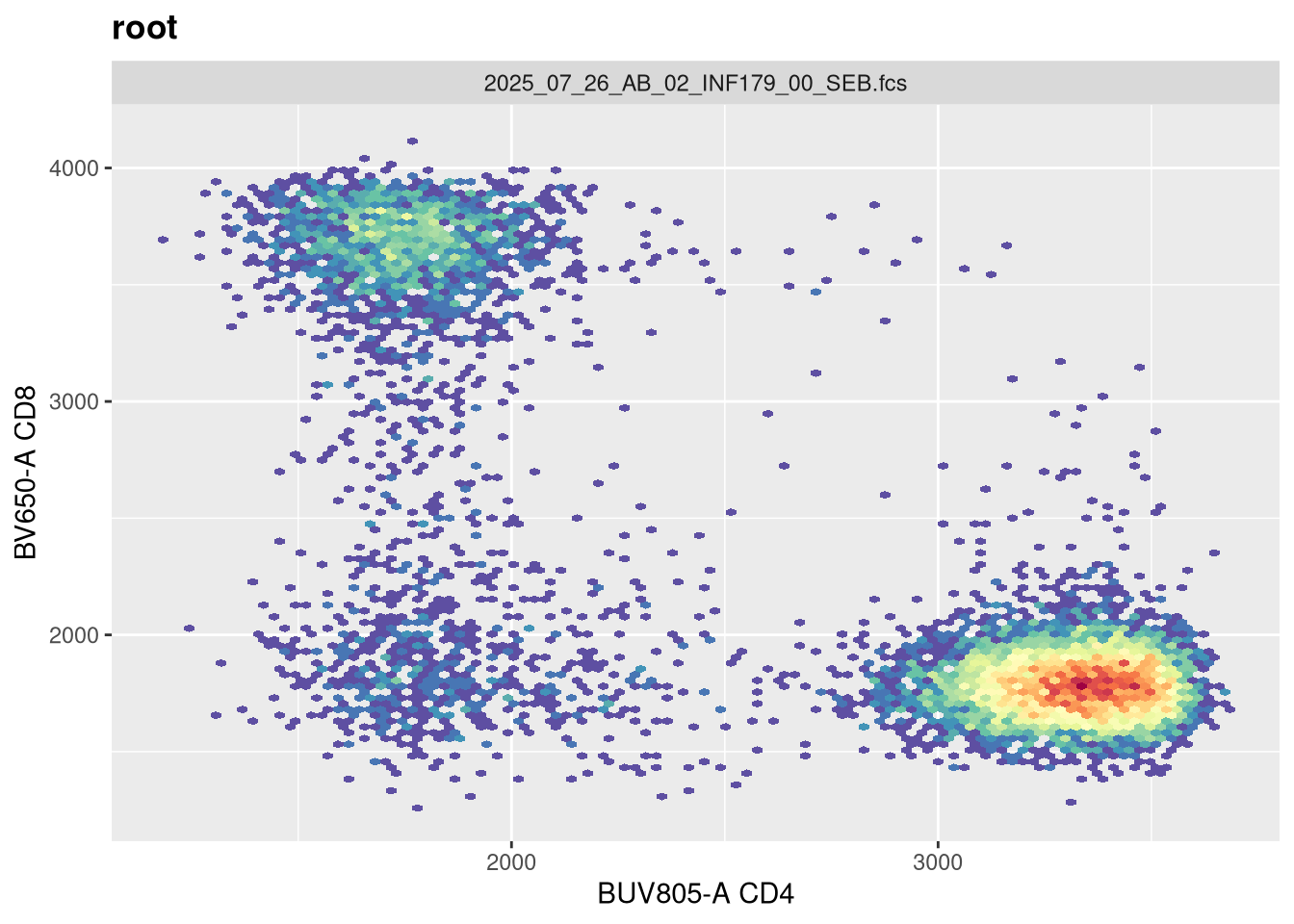
Before we go messing with any of the other setting, let’s tackle width, which tends to be the one that gets missed by the default most often. Normally, in FlowJo most of my plots are set with biexponential transform width of around -1000, so lets go ahead and switch in that value.
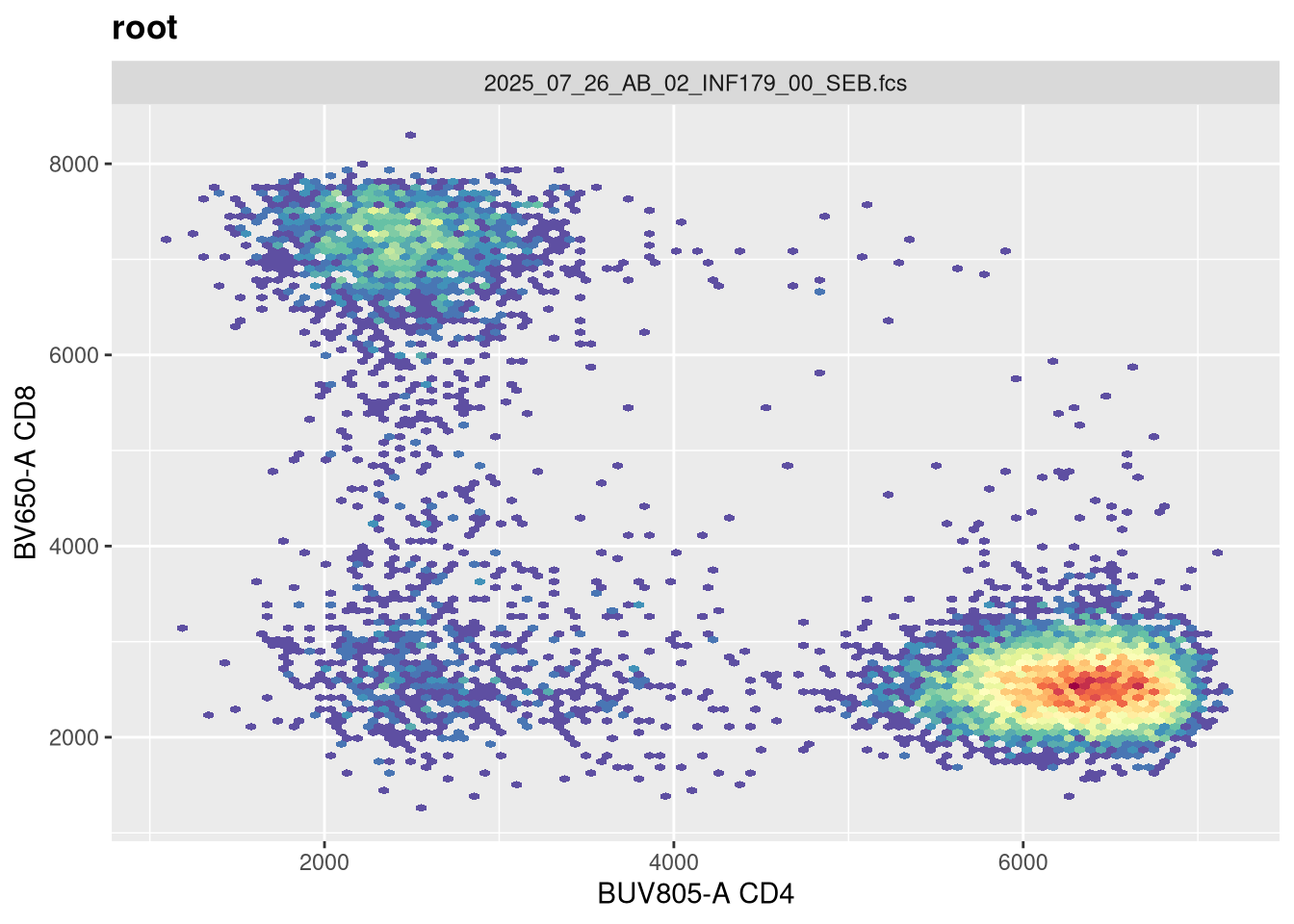
Now that width is set closer to what we would expect, lets alter the next couple arguments and see if they have any effect. The argument channelRange is currently set for 4096. Lets double it:
You will notice, that when we doubled the maxValue, we are phenocopying the result we got when we had width set to -1000, with the CD4 population. This is worth noting. Lets now try instead to reduce it!
And likewise, we are going the opposite to our intended effect. As maxValue can in part be determined by our instrument settings (as well as individual fluorophores), this is an argument we should keep an eye on. Let’s leave it at its current default for now.
pos
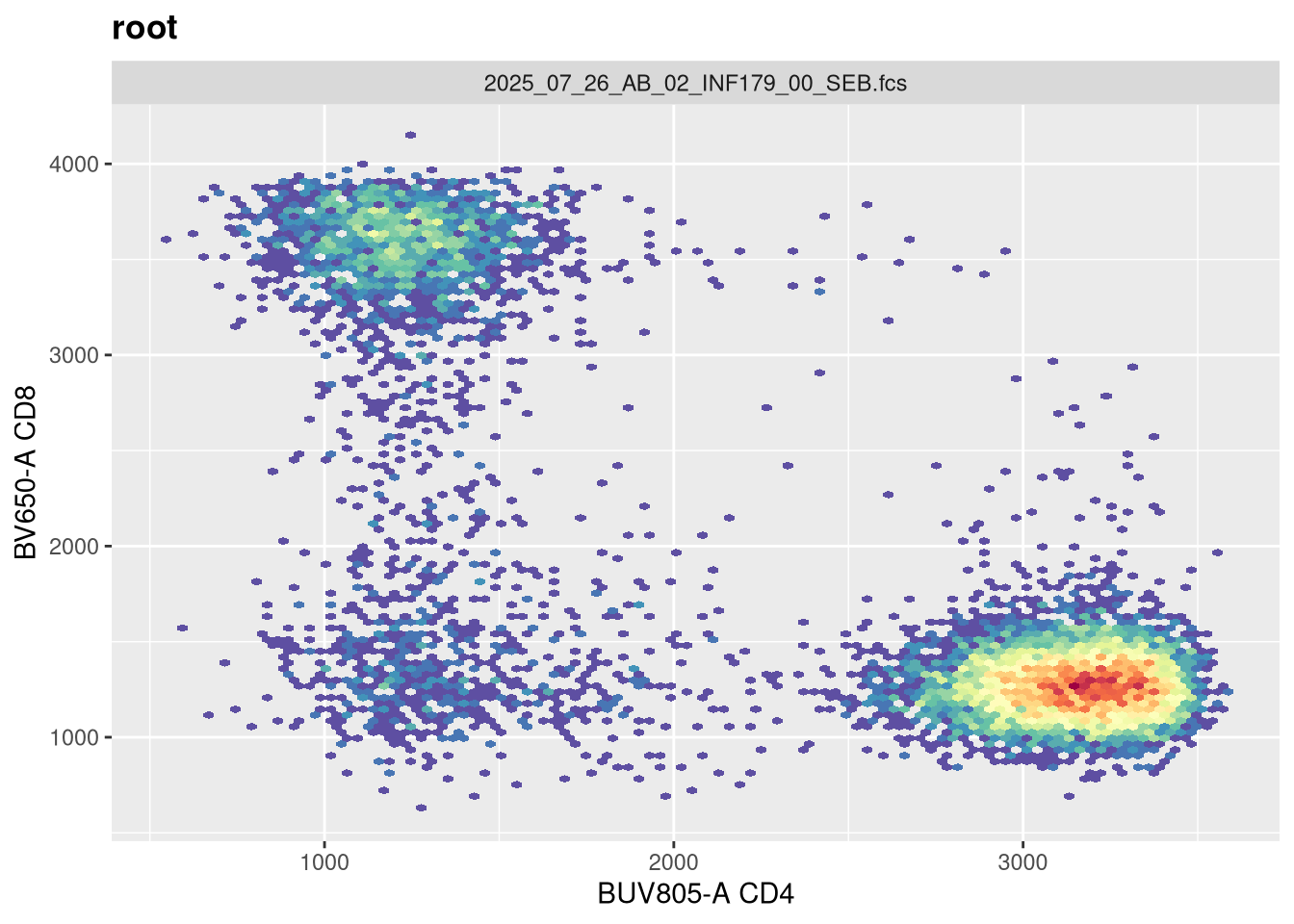
The next argument pos corresponds to the number of positive decades. It is currently set at 4.5
And on first glance no. If we had our settings for 5 positive decades, we would need to adjust both the maxValue and widthBasis to return to a closer to expected shape.
We have looked at how the various arguments can affect the overall visualization, and seen how some of these are interconnected. It might be worthwhile to verify what your own instrument settings are at first (especially in context of maxValue and positive decades), and then use these when starting in R for the first time.
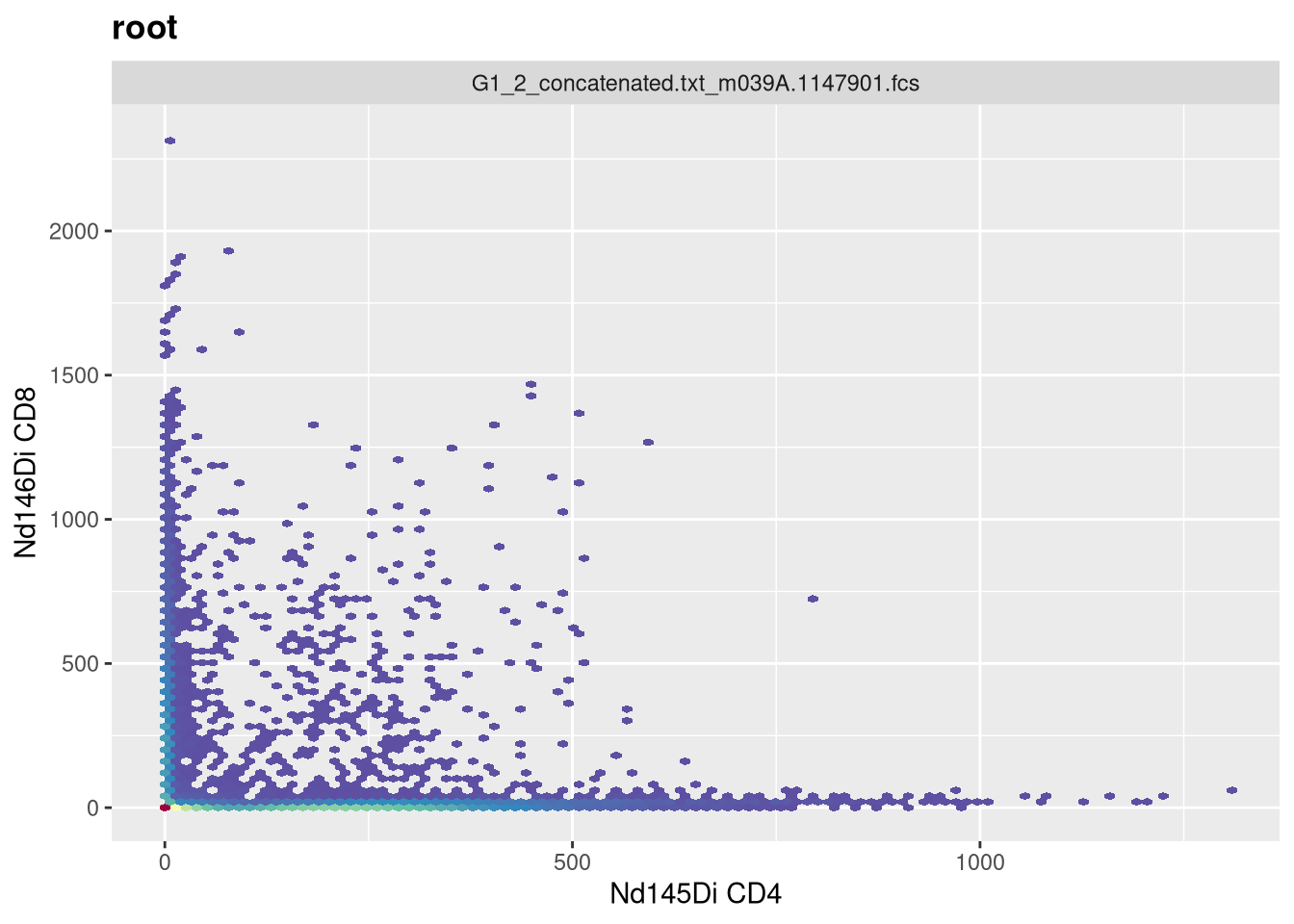
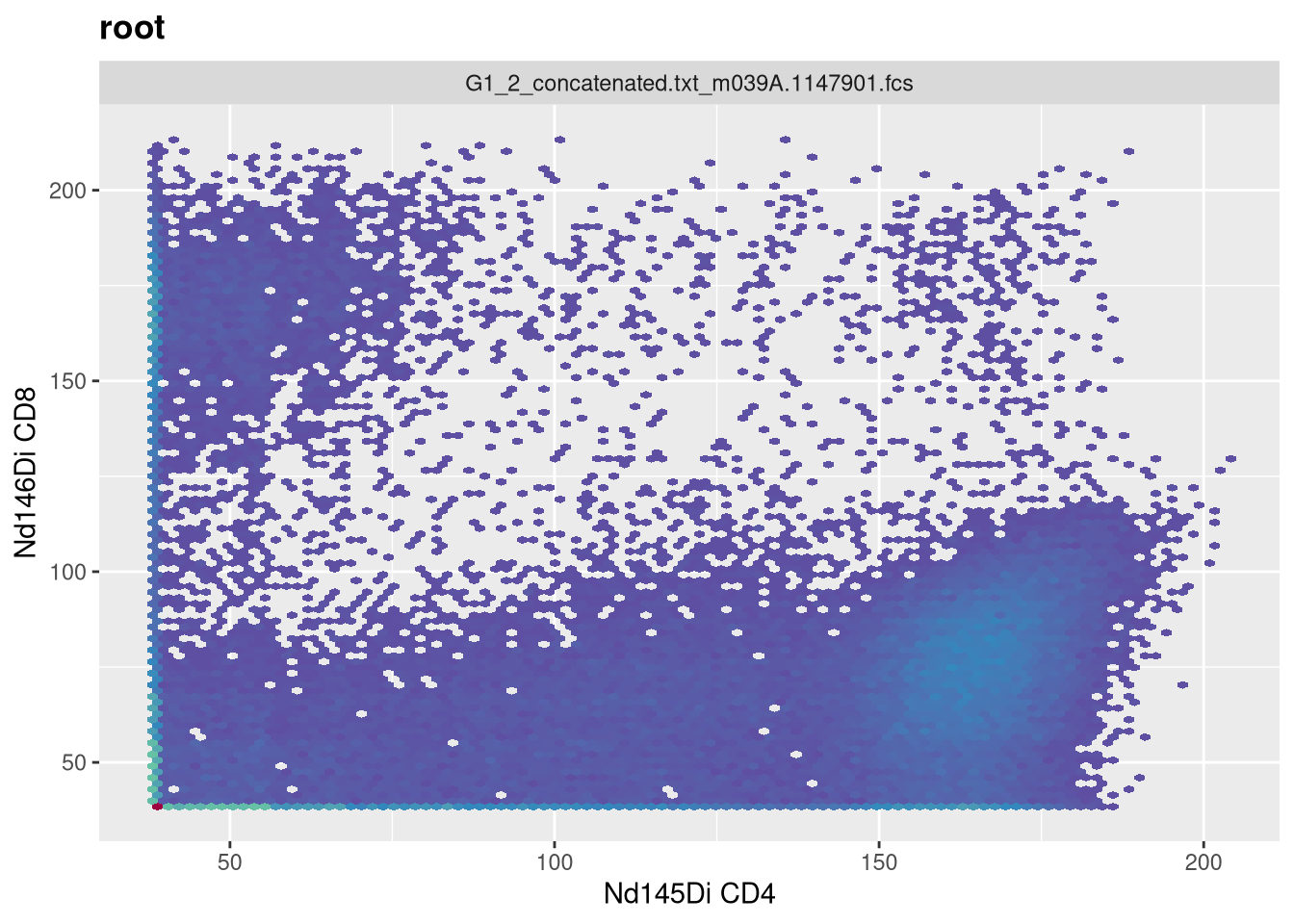
MC
Having now had some experience applying transformations to SFC data, let’s turn and look at how to apply arsinh to our mass cytometry (CyTOF) dataset. Lets once again make sure we have the correct .fcs files
We can then select the particular markers we are interested, that we want to end up transforming. For this example, lets say we wanted to remove the multiple Barcode and Beads entries
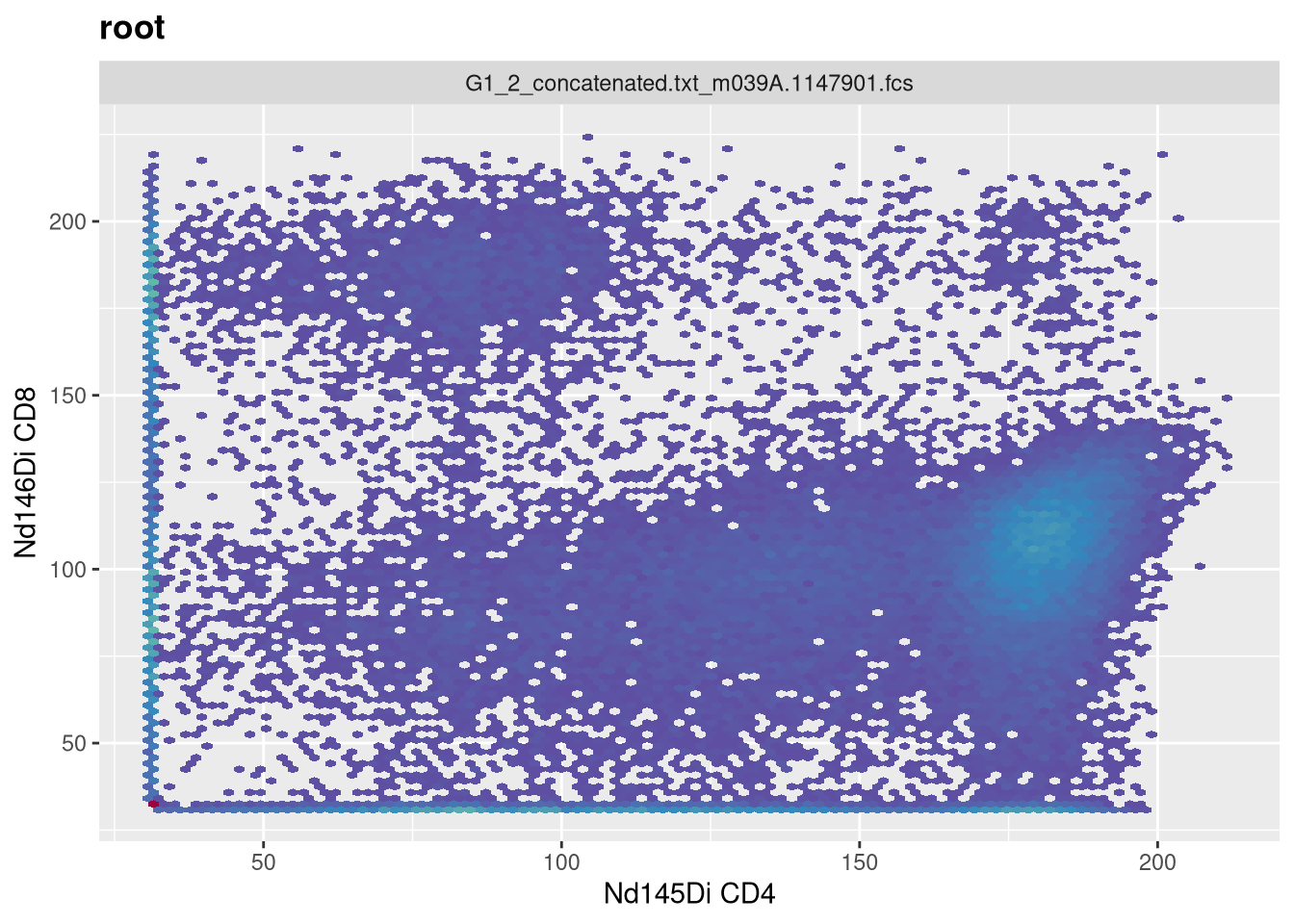
One area where we will separately need to remember whether a transformation is applied or not is later on in the course, when we are trying to extract out the underlying exprs data for use in downstream analysis. If we wanted to do this for our current GatingSet, we first need to fetch the pointer contents to return a CytoSet object.
If you look at the data for a while, you may notice the MFI values seem abnormally abbreviated. This is because the values present were themselves transformed. If we had wanted to retrieve the raw, untransformed values, we would have needed to provide the inverse.transform = TRUE argument when fetching the CytoSet. This would have trigerred the inverse.transformation stored within the transformerList, reverting values back to the original.
Right now, this won’t affect much, but we will encounter where this matters in a couple weeks when we discuss how to visualize fluorescent signatures, and their subsequent use in unmixing.
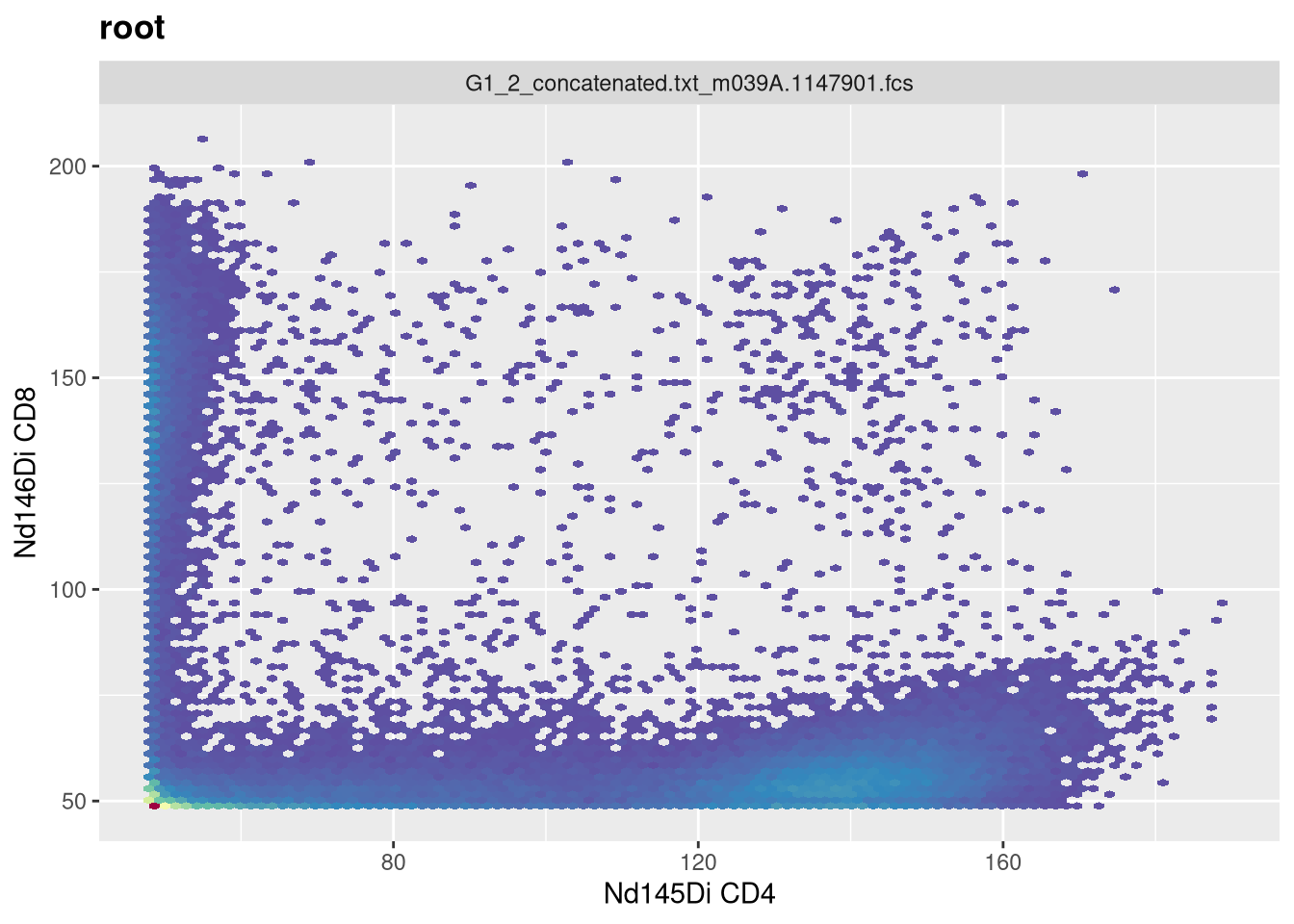
Take Away
This week, we looked at how to set up our underlying data for transformation within the flowWorkspace context, providing the transform and column names of interest to create a transformerList, and then applying it to our GatingSet. We also investigated how to visualize our data and provide additional arguments to ensure that we are optimally transforming the data rather than accepting the default options.
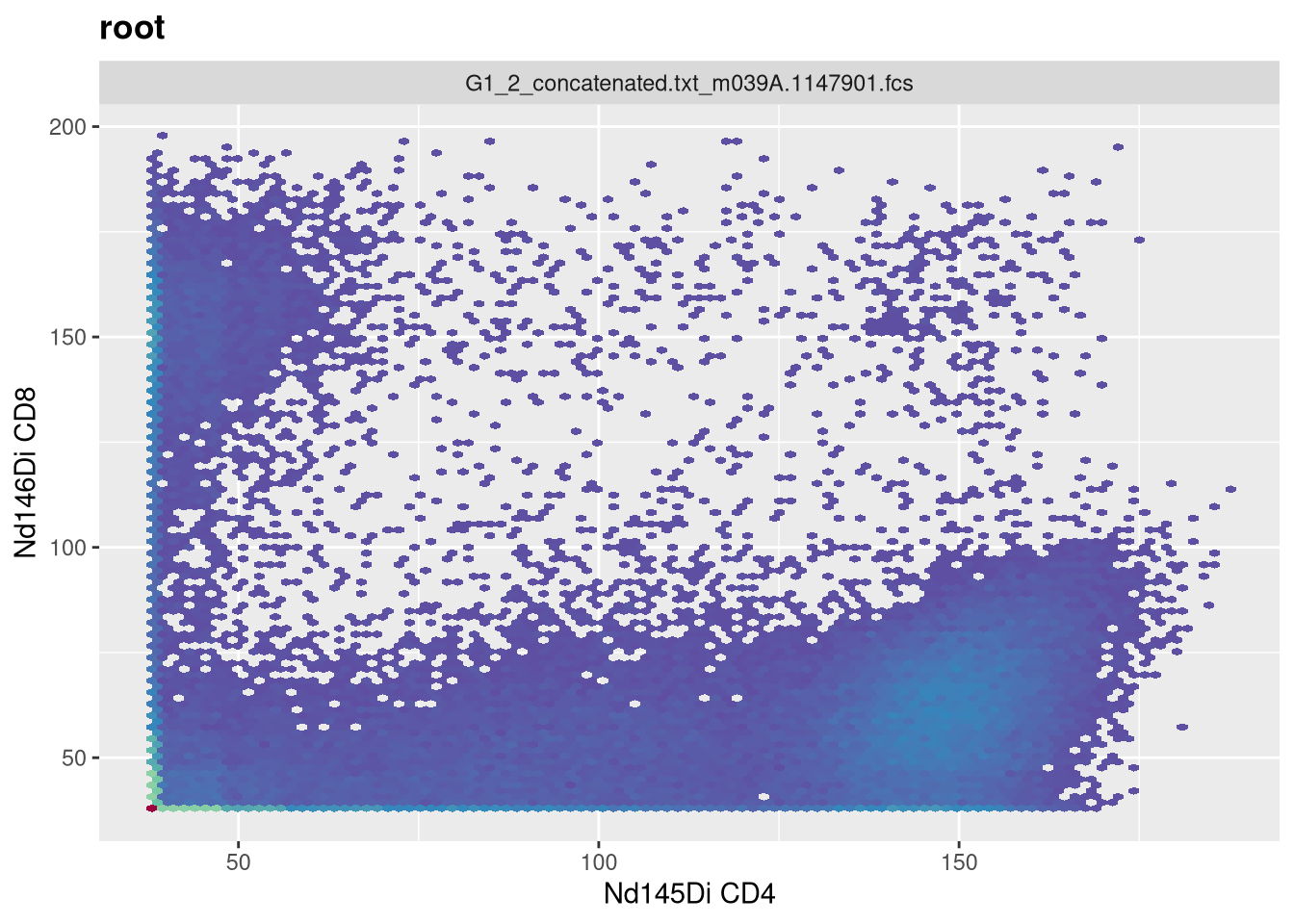
As cytometry instrument platforms will often vary on their range settings, the parameter argument values we used for this particular visualization may not be applicable to your own dataset, so you may need to tinker with your own .fcs files to ensure that you are visualizing the underlying data properly, depending on what you are doing (whether supervised manual analysis, or more unsupervised algorithmic style analysis).
Next week, we will have no class (I will be presenting at the ABRF conference). I had to split this weeks combined Transformation/Compensation lesson into two separate parts, I am still deciding whether to have compensation be a separate stand-alone bonus class (given primary audience is those doing conventional flow cytometry) or to push it into the current rotation ahead of manual and automated gating. I will let you know once I figure it out.
Also, as a heads up, I will be sending out the first class feedback survey. This is our first year doing this course, and any feedback you can provide will help make sure we address any areas that we are falling short to make things better both for those currently taking the course, as well as those who may follow later.
We had not selected FSC and SSC parameters in this attempt, as they are normally displayed in the linear scale. Include them in the list of fluorophores to be transformed, and see how this impacts the visualization (imitating what could accidentally happen in practice if they were left in)
TipProblem 2
For the SFC data, I showed the setup for both Logicle and Biexponential, but didn’t have time to dive into the Logicle transformation. Select a couple markers of interest for the SFC data, visualize and screenshot the before, and then attempt to customize the biexponential arguments to best visualize the underlying data, and then repeat for Logicle. Take screenshots of both and compare/contrast the difference.
TipProblem 3
There are to asinh style transformations provided by the flowWorkspace package. Using the mass cytometry data, select two metal markers of interest, visualize each, customize the arguments until you have properly visualized the underlying populations, and see if you can spot any major differences between the methods.